 Chelsey L. Patten
Chelsey L. Patten Christine E. Cutucache
Christine E. Cutucache- University of Nebraska at Omaha, Omaha, NE, USA
Dozens of murine models of indolent and aggressive B-cell lymphomas have been generated to date. These include those manifesting chronic lymphocytic leukemia (CLL), diffuse large B-cell lymphoma (DLBCL), as well as xenografts of mantle cell lymphoma (MCL). These models have led to an improved understanding of disease etiology, B-cell biology, immunomodulation, and the importance of the tumor microenvironment. Despite these efforts in CLL, DLBCL, and MCL, considerably little progress toward a model of splenic marginal zone lymphoma (SMZL) has been accomplished. Herein, we describe the similarities and differences between CLL, MCL, and SMZL and highlight effective murine models that mimic disease in the two former, in hopes of informing a potential model of the latter. At the time of writing this review, the precise molecular events of SMZL remain to be determined and a treatment regimen remains to be identified. Therefore, based on the efforts put forth in the B-cell lymphoma field throughout the past three decades, the established role of caveolin-1 in B- and T-cell biology as an oncogene or tumor suppressor, and the recurrent deletion or loss of heterozygosity (LOH) of 7q in many cancers, we make recommendations for a murine model of SMZL.
Background
Splenic marginal zone lymphoma (SMZL) is a newly acknowledged cancer that effects primarily middle-aged and elderly patients. This disease primarily effects patients ages 60 and older without evidence of gender predominance (1). Since SMZL is less than a quarter-century old in terms of recognition by the World Health Organization, little progress has been made regarding disease pathogenesis.
Although multiple B-cell lymphoma murine models have been created, currently there is no mouse model dedicated specifically to understanding the disease pathology of SMZL. With no definitive clues as to what drives this disease, the development of a reliable and replicable in vivo model is needed in order to understand the basic oncogenic factors leading to SMZL. In this review, we discuss possible contributing factors in tumorigenesis, highlight three similar B-cell malignancies [i.e., chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), and SMZL]—including their shared immunophenotype, as well as currently existing murine models of these neoplasms—and finally we discuss the role for 7q in SMZL. Ultimately, we make a recommendation for the generation of a murine model with a knockout at 7q involving caveolin-1 (CAV1), based on its role in related B-cell malignancies.
Possible Contributing Factors Leading to Tumorigenesis
When designing a mouse model that can accurately recapitulate SMZL, it is imperative to understand all of the factors that may contribute to the tumorigenesis of the disease. To shed light on which factors specifically contribute to the occurrence of SMZL, a population-based study was conducted in the United States on a cohort from 2001–2008 on SMZL incidence and patient survival. Several possible contributing factors emerged from this study, including autoimmune disease (20% of cases), environmental factors, and aging-related effects, such as chronic inflammation, DNA damage, and a diminished immune response (1, 2). In addition to these, specific factors that might influence oncogenic events can include regulatory elements, infection, genetic mutations (transcriptomic), or epigenetics (methylation patterns).
Differences in disease drivers can distinguish SMZL into two different subtypes. Approximately half of SMZL cases are likely caused by infectious disease, more specifically, the hepatitis C virus [HCV; (3)]. This infectious disease-driven subtype suggests that virally transformed animal models might be an effective way to study disease onset and progression. However, chromosomal aberrations are present in the vast majority (>70%) of all SMZL cases, implying that a genetic component may serve as the major driver of tumorigenesis (4). Clinically, these two subtypes of SMZL are distinct in terms of disease progression and prognosis for patients.
Infections
Many studies suggest an antigenic role for tumorigenesis of SMZL. In fact, 30–40% of cases have shown to be disease driven (5). The possible causative role of HCV has been suggested both based on high prevalence as well as multiple epidemiologic and therapeutic studies (1, 3, 6). The etiology of HCV and its relation to B-cell lymphomas in general has been correlated to the geographic distribution in a few studies with small case numbers, mainly in Japan and Southern Italy. Overall, local HCV prevalence along with genetic and environmental factors may play a part in the geographically diverging results (3). Although geographic distribution has not yet been shown to have a significant impact on prevalence rates, an increase in HCV-positive persons from 2.7 million to 3.2 million from 1994–1998 to 1999–2002 mirrored increased prevalence rates of SMZL during this same time frame (2). Furthermore, from 2001 to 2008, the incidence of SMZL continued to steadily climb (2) and now comprises 2% of all lymphoid neoplasms (7). Although the exact pathogenesis mechanism is unknown, both chronic antigen stimulation and viral lymphotropism may contribute to progression of the malignant cell (3).
Findings that intraclonal diversification caused by ongoing somatic hypermutation was identified in 81% of the rearrangements using the IGHV1-2*04 genes versus only 40% of rearrangements using other IGHV genes. This supports the idea of antigen selection in SMZL ontogeny, as well as the possibility of ongoing antigen involvement throughout the progression of the disease, even possibly toward diffuse large B-cell lymphoma (DLBCL) like suggested in the “Multistep Theory of Lymphomagenesis” (7–10). The 14q32 band holds IGHV, and translocations involving the 14q32 band have been detected less frequently in SMZL than in non-Hodgkin lymphomas. Half of SMZL patients carry an increased load of IGHV somatic mutations, which is associated with improved prognosis (11). Combined, this supports the argument that IGHV1-2*04 SMZL is, in fact, a distinct molecular SMZL subtype that needs to be recognized and studied in vivo (8–11).
Genetic Mutations
Although many SMZL cases have shown a possibility for a disease-driven etiology, more than 70% of SMZL cases show some form of chromosomal aberration, most predominantly a loss of heterozygosity (LOH) at 7q (2). This 7q LOH is observed in 40–50% of total cases, and the 7q31–32 deletion is present in approximately 45% of all cases, serving as the most common cytogenetic abnormality, thus suggesting a genetic driver (8, 9, 12–16). Many studies have been done in hopes of finding exactly which genes and pathways are altered in SMZL, therefore serving as potential targets on the development of SMZL murine models. A summary of these studies can be found in Table 1 (8, 9, 11, 13–15, 17–25). Although many genes (most notably NOTCH2, KLF2, KLF4, and BIRC3) have been reported as mutated in SMZL, we suggest that the 7q deletion is of primary importance, as it is possible this deletion serves as a marker for disease progression and may even be a causative event, rather than a pro-survival function as was previously speculated (9).
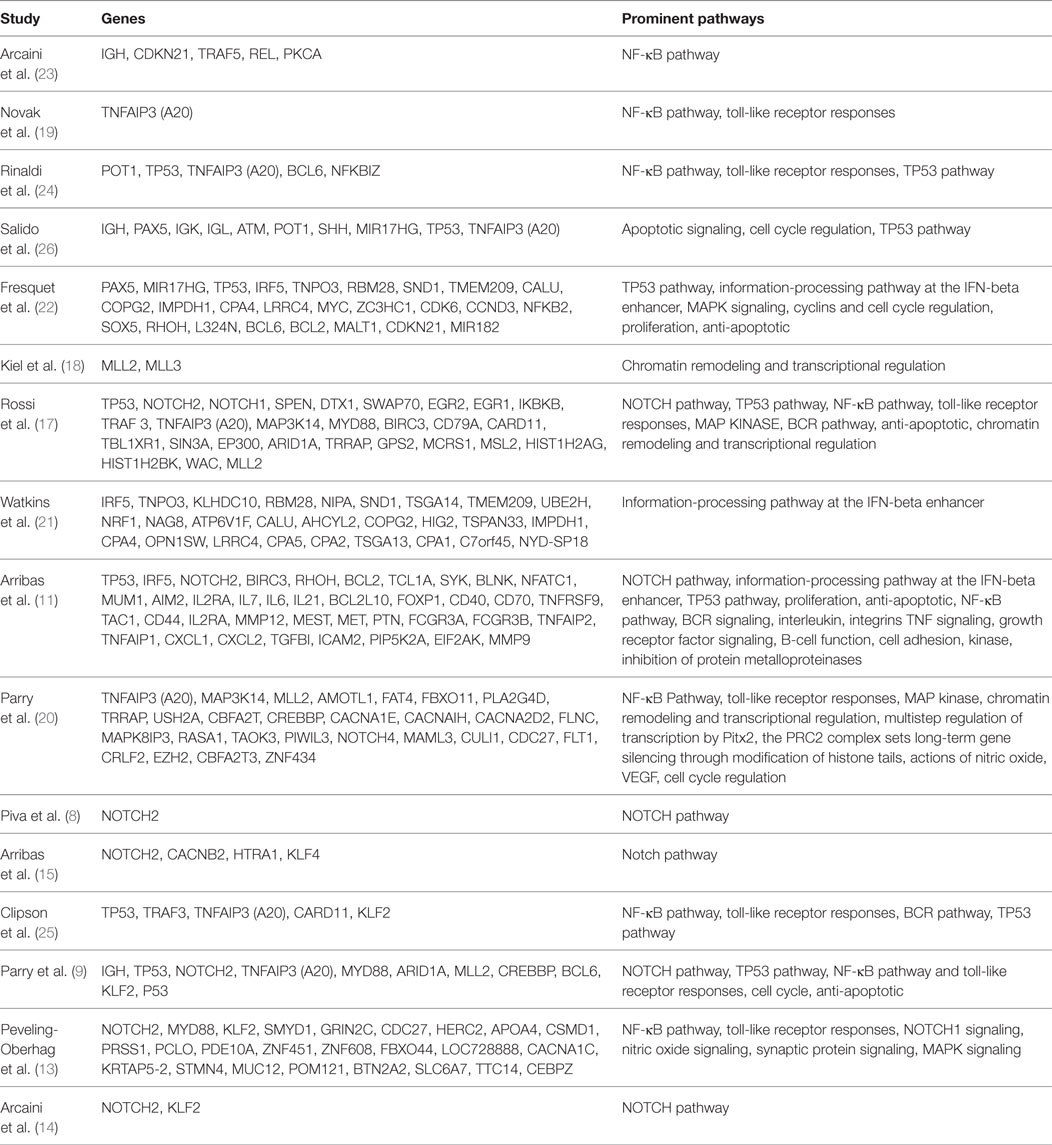
Table 1. Summary of reported genomic mutations or deletions in splenic marginal zone lymphoma (SMZL) (8, 9, 11, 13–15, 17–25).
Methylation Patterns
In a genome-wide DNA-promoter methylation profiling study by Arribas et al., two main clusters were distinguished based on the degree of promoter DNA methylation (15). This high-M cluster not only had an inferior outcome and showed high risk for histologic transformation to DLBCL but suggests that DNA hypermethylation could act together with 7q31-32 deletion, NOTCH2 mutation, and IGHV1-02, to determine a distinct genetic and epigenetic subgroup of SMZL (11).
Birds of a Feather: B-Cell Neoplasms CLL, MCL, and SMZL
Chronic lymphocytic leukemia, MCL and SMZL are all neoplasms that affect mature B-cells; yet, in contrast to its B-cell counterparts, SMZL is the least studied. These three malignancies have major commonalities (27–30), including some shared surface markers, shared pathways for disease progression (15), and hypothesized infectious disease drivers, including HCV (Figure 1).
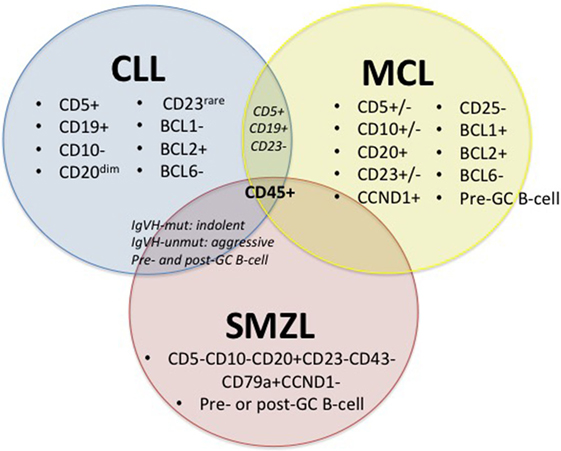
Figure 1. Diagnostic factors of CLL, MCL, and SMZL. +/−, rarely expressed; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; SMZL, splenic marginal zone lymphoma (27–30).
Figure 1 depicts immunophenotypical similarities and differences across CLL, MCL, and SMZL. The expression of CD5, CD19, BCL2, and rarely CD23 along with the absence of CD10 and BCL1 with diminished CD20 set CLL apart from its close relatives. However, the presence of CD5 and CD19 and the absence of CD23 can also indicate MCL, so it is important to run a multitude of panels in order to solidify a diagnosis. MCL can display variable expression of CD5, CD10, and CD23 and express CD20, CCND1, BCL1, and BCL2, while lacking CD25 and BCL6. SMZL expresses CD20 and CD79a but lacks CD5, CD10, CD23, CD43, and CCND1. Both CLL and SMZL are thought to originate from pre- or post-germinal center B-cells. This etiology differs from that of MCL, which is known to arise from pre-germinal center B-cells. Last, CLL and SMZL have a common prognostic factor with mutated IGVH, leading to improved outcome for patients (27–30).
Existing Murine Models
The development and maintenance of murine models are important due to (i) difficulty in gaining access to patient samples, (ii) inability to study host:tumor interactions in an ex vivo system, and (iii) lack of cell lines that adequately recapitulate human disease (31). Both CLL and MCL have been well-characterized, including having murine models developed, while a dedicated, specific SMZL murine model driven by a transgene is still absent. Although previous SMZL models have been attempted, they often lead to development of DLBCL and, therefore, do not serve as true SMZL models (since clinically when SMZL progresses to DLBCL, additional transcriptomic events occur). The current murine models for CLL and MCL, as well as those identified to-date for SMZL are discussed below.
Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia is the most common adult leukemia in the Western world, and consequently, in comparison to its B-cell counterparts, murine models for CLL are the most well-defined. This is in part due to their ability to accurately recapitulate human disease, specifically the tumor microenvironment that is paramount for the maintenance of CLL cells. Because of this, it is imperative that CLL must harbor in supportive organs both in patients and in mouse models (31, 32). The most accurate recapitulation of CLL in vivo is arguably the Eμ-TCL1 transgenic mouse from the C. Croce lab (31), and this argument is emphasized in a review by Bresin et al. (33). Conversely, the NZB-IRF4−/− model from R. Lu’s lab is reminiscent of the aggressive phenotype of CLL (34). The NZB-IRF4−/− mice developed spontaneous CLL at 100% penetrance – thereby indicating a relationship between levels of IRF4 and CLL development. In these mice, CLL cells with VH11 proliferated predominantly in the spleen, thus allowing this model to serve as a successful study agent for molecular pathogenesis as well as for therapeutic techniques (35).
While the Eμ-TCL1 and NZB-IRF4−/− lines most accurately recapitulate human CLL, cell line xenografts have also been established. Kellner and colleagues developed the first long-term proliferative cell line for CLL from a (del)17p case and furthered this development by using this cell line to create a xenograft model that recapitulates CLL (31, 36). These are major strides in CLL research, as no other group has been able to create a reliable, actively replicating CLL cell line. Other examples include xenografts of peripheral blood CLL and MCL cells in alymphoid mice by other labs (37, 38) thoroughly reviewed by Chen and Chiorazzi (39).
Not surprisingly, as CLL is the most common adult leukemia and receives the greatest amount of work by clinicians and scientists alike, major strides have been made to create reliable murine models for disease (including both the indolent and aggressive phenotypes).
Mantle Cell Lymphoma
Mantle cell lymphoma is an aggressive, yet rare, neoplasm that affects less than 200,000 people in the United States annually (10). In comparison to CLL cells, MCL cells are far less reliant on tumor microenvironment. Therefore, mouse models of MCL have largely come from xenografts that poorly mimic tumor microenvironmental conditions; however, MCL cells are less reliant on tumor microenvironment than CLL cells. By utilizing the method of tail vein injection, in vivo models of metastatic human MCL were established as described herein, briefly. Both primary cell lines from MCL cases and MCL cell lines (including Jeko-1, Mino, Rec-1, Hbl-2, and Granta-519) were used to generate disease in immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wgl/Szj mice or NOD.SCID mice (40–42). Hegde et al. (41) and Ahrens et al. (40) in the S. S. Joshi’s lab created therapy-resistant models to study the impact of novel adjuvants for improving patient outcome. These xenografts were effective; namely, 7 out of 12 patient samples allowed for engraftment observation with the use of primary MCL cell lines, while all 5 of the MCL cell lines showed tumor engraftment. The overall survival of these mice varied with regards to the specific cell line used but ranged from 22 ± 1 to 54 ± 3 days (41, 42). Furthermore, the NOD.Cg-Prkdcscid Il2rgtm1Wgl/Szj MCL mouse model has been shown to aid in testing systemic chemotherapy, monoclonal antibodies, and angiogenesis inhibitors, which will serve as a significant tool in understanding this disease and advancing diagnostic and therapeutic techniques. Moreover, the NOD/SCID IL2Rgamma-null xenograft model was effective for studying p53-mutated CLL and ATM-mutated MCL (38).
Taken together, these studies have led to recapitulation of MCL (both from primary cells and cell lines) in vivo, thereby providing effective murine models to use for drug testing as well as to increase the understanding of basic disease biology and disease progression.
Splenic Marginal Zone Lymphoma
Like MCL, SMZL is a rare neoplasm. Due to its relatively recent identification, it is not surprising that many questions linger about its oncogenesis and pathogenesis. SMZL has been observed spontaneously in a few murine models, namely, NFS.N mice expressing murine leukemia virus (5, 43, 44) as well as a transcription factor-induced transgenic line with murine leukemia virus (45). Although the latter model develops SMZL, it quickly progresses into full-blown DLBCL. Similarly, the loss of a potent tumor suppressor, p53, leads to a SMZL-like phenotype in mice (46). All of these mouse models aiming to recapitulate human SMZL in vivo include a viral component or a major genomic hit that is non-reminiscent of the human presentation.
An in vivo murine model to mimic SMZL remains to be created, but because of its potential at assisting hematologists in understanding basic B-cell biology, germinal center reactions, and ultimately tumorigenesis, a new model is most certainly warranted. The recent identification of the role for the 7q31-32 region, a common minimally deleted region in human cancers, reignites the efforts to pursue causative mutations in a coding or non-coding region within that locus.
LOH AT 7q31: Potential for Murine Model Development to Mimic SMZL
There are many regions of the genome considered “fragile” sites, and the long arm of chromosome 7 is one such region. Some studies have mapped the specific portions of this region that show causality with tumorigenesis, but others have simply recognized the region as a whole. LOH is observed at 7q31-34 in the following cancers: SMZL, meningioma, papillary thyroid carcinoma, ovarian cancer, acute myeloid leukemia, myelodysplastic syndrome, and pancreatic carcinoma. Deletions at 7q have been reported in a variety of human neoplasias including leukemia, breast, ovary, colon, prostate, gastric, head and neck, pancreatic, and renal cell carcinomas (47).
Several groups have sought a “master regulator” within the commonly deleted 7q region (and corresponding downregulated genes) in severe cases of SMZL. However, despite valiant efforts at well-controlled experiments (9), a master regulator has yet to be identified. The development of such a model to explore driver mutations leading to SMZL would shed light on SMZL pathophysiology as well as biological behavior, thereby serving as an effective model.
A Role for Cav1
Fragile site FRA7G holds two tumor suppressor genes, including CAV1 and TES. Cav1 has been shown to serve as an oncogene or tumor suppressor depending on cell or tissue type, microenvironmental influence, or disease progression (48). CAV1 7q31.2 deletions/translocations appear to be downregulated in ovarian, lung, and mammary tumors while upregulated in prostate, bladder, thyroid, and esophageal carcinomas (47). Haploinsufficiency of CAV1 is enough to induce partial transformation of human breast epithelial cells (49). Additionally, the loss of Cav1 combined with MMTV-PyMT leads to the acceleration of mammary tumors (47). Cav1-knockout mice do not spontaneously develop tumors, although the skin is more susceptible to chemical carcinogenic treatment. Specifically, when Cav1−/− mice were exposed to 7,12-dimethylbenz(a) anthracene, epidermal cells significantly upregulated CCND1 and ERK1/2 and mice developed hyperplastic ductal epithelium (47, 50).
In addition to carcinogenic challenges in Cav1-null mice, recent studies identify a link between Cav1 and both B- and T-cell neoplasms. Specifically, CAV1 was identified as a major immunoregulator, which was significantly upregulated in aggressive CLL cases (48). When Eμ-TCL1-Tg-Cav1−/− mice were generated, they displayed a significantly more aggressive phenotype than the indolent Eμ-TCL1 model (51). Moreover, the role of Cav1 is well-documented in immune synapse formation (48, 52), thereby suggesting another contribution to immune evasion and tumorigenesis. Most recently reported, CAV1 is deleted in up to 45% of SMZL cases (16). Additionally, CAV1 has been documented to play a key role as a potential diagnostic marker in T-cell malignancies as it is significantly upregulated in more than 67% of T-cell malignancies (53). Taken together, these data suggest a clear role for CAV1 in both B- and T-cell neoplasms. Therefore, as CAV1 is lost or mutated in up to half of all SMZL cases and the 7q region (where CAV1 is located) is commonly deleted, we suggest that a murine model under an oncogenic driver, coupled with Cav1 LOH, could be an effective model.
Summary
CAV1 resides at the 7q region and is heterogeneously expressed in cancer. Specifically, CAV1 is downregulated in SMZL cases, while upregulated in aggressive cases of chronic lymphocytic leukemia and five types of mature T-cell lymphomas. We (and others) have demonstrated a critical role for CAV1 in immune synapse formation, cellular proliferation, and cellular migration. Cav1−/− mice are immunosuppressed, but the mechanism by which this immune dysregulation occurs is currently unknown. Improving our understanding of the basic biology of CAV1 in the immune response will translate to (i) determining its role in the immune response governing antigen-presentation events in healthy and diseased immune responses and (ii) how CAV1 controls immunophenotype/immune cell frequency. We have shown CAV1 to be significantly dysregulated across more than 67% of cases of mature T-cell lymphomas as well as in aggressive cases of chronic lymphocytic leukemia. Recent data from others identify the 7q deletion in another mature B-cell lymphoma: SMZL. Therefore, results generated from this proposal would be applicable across lymphoid neoplasms. Taken together, our preliminary data support a hypothesized role for CAV1 in lymphoid tumor progression, likely through an immune regulatory method.
Based on the knockout studies involving p53, coupled with the high incidence of spontaneous Hodgkin’s lymphoma development on the Swiss Jim Lambert mice, we suggest the development of a SMZL murine model with Cav1 LOH with or without an oncogenic driver, such as p53−/− or MYC-Tg. Finally, the CAV1 deletion being present in up to 45% of all SMZL cases suggests that generating a MYC-Tg-Cav1−/− mouse could serve as a more representative clinical mimic of SMZL. Based on the known role of CAV1 in aggressive cases of both B- and T-cell malignancies, its role in immune regulation through immune synapse formation, its role in the tumor microenvironment, and the frequency of LOH at 7q, we suggest that this deletion could serve as a marker for disease progression and may even be a causative event.
Author Contributions
Both CP and CC conceptualized, wrote, and edited the manuscript and approved this final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors acknowledge the Department of Biology and the College of Arts and Sciences at the University of Nebraska at Omaha and the University of Nebraska Foundation for supporting this work.
References
1. Matutes E, Oscier D, Montalban C, Berger F, Callet-Bauchu E, Dogan A, et al. Splenic marginal zone lymphoma proposals for a revision of diagnostic, staging and therapeutic criteria. Leukemia (2008) 22:487–95. doi: 10.1038/sj.leu.2405068
2. Liu L, Wang H, Chen Y, Rustveld L, Liu G, Du XL. Splenic marginal zone lymphoma: a population-based study on the 2001–2008 incidence and survival in the United States. Leuk Lymphoma (2013) 54(7):1380–6. doi:10.3109/10428194.2012.743655
3. Sulyok M, Makara M, Újhelyi E, Vályi-Nagy I. Non-Hodgkin lymphoma and hepatitis C: where we are and what next? Pathol Oncol Res (2014) 21:1–7. doi:10.1007/s12253-014-9845-z
4. Baliakas P, Strefford JC, Bikos V, Parry M, Stamatopoulos K, Oscier D. Splenic marginal-zone lymphoma: ontogeny and genetics. Leuk Lymphoma (2015) 56(2):301–10. doi:10.3109/10428194.2014.919636
5. Franco V, Florena AM, Iannitto E. Splenic marginal zone lymphoma. Blood (2003) 101:2464–72. doi:10.1182/blood-2002-07-2216
6. Vannata B, Stathis A, Zucca E. Management of the marginal zone lymphomas. Cancer Treat Res (2015) 165:227–49. doi:10.1007/978-3-319-13150-4_9
7. [SEER] Surveillance, Epidemiology, and End Results Program. National Institutes of Health. (2016). Available from: http://seer.cancer.gov/seertools/hemelymph/51f6cf57e3e27c3994bd5327/
8. Piva R, Deaglio S, Famà R, Buonincontri R, Scarfò I, Bruscaggin A, et al. The Krüppel-like factor 2 transcription factor gene is recurrently mutated in splenic marginal zone lymphoma. Leukemia (2014) 29(2):503–7. doi:10.1038/leu.2014.294
9. Parry M, Rose-Zerilli MJ, Ljungström V, Gibson J, Wang J, Walewska R, et al. Genetics and prognostication in splenic marginal zone lymphoma: revelations from deep sequencing. Clin Cancer Res (2015) 21(18):4174–83. doi:10.1158/1078-0432.CCR-14-2759
10. Bikos V, Karypidou M, Stalika E, Baliakas P, Xochelli A, Sutton LA, et al. An immunogenetic signature of ongoing antigen interactions in splenic marginal zone lymphoma expressing IGHV1-2*04 receptors. Clin Cancer Res (2016) 22(8):2032–40. doi:10.1158/1078-0432.CCR-15-1170
11. Arribas JA, Gómez-Abad C, Sánchez-Beato M, Martinez N, DiLisio L, Casado F, et al. Splenic marginal zone lymphoma: comprehensive analysis of gene expression and miRNA profiling. Mod Pathol (2013) 26(7):889–901. doi:10.1038/modpathol.2012.220
12. Tsieh S. Flow Cytometry, Immunohistochemistry, and Molecular Genetics for Hematologic Neoplams. Philadelphia: Lippincott Williams & Wilkins, a Wolters Kluwer Business (2012).
13. Peveling-Oberhag J, Wolters F, Döring C, Walter D, Sellmann L, Scholtysik R, et al. Whole exome sequencing of microdissected splenic marginal zone lymphoma: a study to discover novel tumor-specific mutations. BMC Cancer (2015) 15:773. doi:10.1186/s12885-015-1766-z
14. Arcaini L, Rossi D, Paulli M. Splenic marginal zone lymphoma: from genetics to management. Blood (2016) 127(17):2072–81. doi:10.1182/blood-2015-11-624312
15. Arribas AJ, Rinaldi A, Mensah AA, Kwee I, Cascione L, Robles EF, et al. DNA methylation profiling identifies two splenic marginal zone lymphoma subgroups with different clinical and genetic features. Blood (2015) 125(12):1922–31. doi:10.1182/blood-2014-08-596247
16. Ruiz-Ballesteros E, Mollejo M, Rodriguez A, Camacho FI, Algara P, Martinez N, et al. Splenic marginal zone lymphoma: proposal of new diagnostic and prognostic markers identified after tissue and cDNA microarray analysis. Blood (2005) 106(5):1831–8. doi:10.1182/blood-2004-10-3898
17. Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med (2012) 209(9):1537–51. doi:10.1084/jem.20120904
18. Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med (2012) 209(9):1553–65. doi:10.1084/jem.20120910
19. Novak U, Rinaldi A, Kwee I, Nandula SV, Rancoita PM, Compagno M, et al. The NF-{kappa}B negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood (2009) 113(20):4918–21. doi:10.1182/blood-2008-08-174110
20. Parry M, Rose-Zerilli MJ, Gibson J, Ennis S, Walewska R, Forster J, et al. Whole exome sequencing identifies novel recurrently mutated genes in patients with splenic marginal zone lymphoma. PLoS One (2013) 8(12):e83244. doi:10.1371/journal.pone.008324
21. Watkins AJ, Hamoudi RA, Zeng N, Yan Q, Huang Y, Liu H, et al. An integrated genomic and expression analysis of 7q deletion in splenic marginal zone lymphoma. PLoS One (2012) 7(9):e44997. doi:10.1371/journal.pone.0044997
22. Fresquet V, Robles EF, Parker A, Martinez-Useros J, Mena M, Malumbres R, et al. High-throughput sequencing analysis of the chromosome 7q32 deletion reveals IRF5 as a potential tumour suppressor in splenic marginal-zone lymphoma. Br J Haematol (2012) 158(6):712–26. doi:10.1111/j.1365-2141.2012.09226.x
23. Arcaini L, Lazzarino M, Colombo N, Burcheri S, Boveri E, Paulli M, et al. Splenic marginal zone lymphoma: a prognostic model for clinical use. Blood (2006) 107(12):4643–9. doi:10.1182/blood-2005-11-4659
24. Rinaldi A, Mian M, Chigrinova E, Arcaini L, Bhagat G, Novak U, et al. Genome-wide DNA profiling of marginal zone lymphomas identifies subtype-specific lesions with an impact on the clinical outcome. Blood (2010) 117(5):1595–604. doi:10.1182/blood-2010-01-264275
25. Clipson A, Wang M, de Leval L, Ashton-Key M, Wotherspoon A, Vassiliou G, et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia (2015) 29(5):1177–85. doi:10.1038/leu.2014.330
26. Salido M, Baro C, Oscier D, Stamatopoulos K, Dierlamm J, Matutes E, et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B-cell lymphomas: a multicenter study of the Splenic B-cell Lymphoma Group. Blood (2010) 116(9):1479–88. doi:10.1182/blood-2010-02-267476
27. Matutes E, Morilla R, Owusu-Ankomah K, Houlihan A, Catovsky D. The immunophenotype of splenic lymphoma with villous lymphocytes and its relevance to the differential diagnosis with other B-cell disorders. Blood (1994) 83(6):1558–62.
28. Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s lymphoma classification project. J Clin Oncol (1998) 16(8):2780–95.
29. Savilo E, Campo E, Mollejo M, Pinyol M, Piris MA, Zukerberg LR, et al. Absence of cyclin D1 protein expression in splenic marginal zone lymphoma. Mod Pathol (1998) 11(7):601–6.
30. Ghia P, Guida G, Stella S, Gottardi D, Geuna M, Strola G, et al. The pattern of CD38 expression defines a distinct subset of chronic lymphocytic leukemia (CLL) patients at risk of disease progression. Blood (2003) 101(4):1262–9. doi:10.1182/blood-2002-06-1801
31. Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A (2002) 99:6955–60. doi:10.1073/pnas.102181599
32. Kurtova AV, Balabrishnan K, Chen R, Ding W, Schnabl S, Quiroga MP, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood (2009) 114:4441–50. doi:10.1182/blood-2009-07-233718
33. Bresin A, D’Abundo L, Narducci MG, Fiorenza MT, Croce CM, Negrini M, et al. TCL1 transgenic mouse model as a tool for the study of therapeutic targets and microenvironment in human B-cell chronic lymphocytic leukemia. Cell Death Dis (2016) 7:e2071. doi:10.1038/cddis.2015.419
34. Ma S, Shukla V, Fang L, Gould KA, Joshi SS, Lu R. Accelerated development of chronic lymphocytic leukemia in New Zealand Black mice expressing a low level of interferon regulatory factor 4. J Biol Chem (2013) 288(37):26430–40. doi:10.1074/jbc.M113.475913
35. Shukla V, Ma S, Hardy RR, Joshi SS, Lu R. A role for IRF4 in the development of CLL. Blood (2013) 122(16):2848–55. doi:10.1182/blood-2013-03-492769
36. Kellner J, Wierda W, Shpall E, Keating M, McNiece I. Isolation of a novel chronic lymphocytic leukemic (CLL) cell lines and development of an in vivo mouse model of CLL. Leuk Res (2016) 40:54–9. doi:10.1016/j.leukres.2015.10.008
37. Oldreive CE, Skowronska A, Davies NJ, Parry H, Agathanggelou A, Krysov S, et al. T-cell number and subtype influence the disease course of primary chronic lymphocytic leukaemia xenografts in alymphoid mice. Dis Model Mech (2015) 8(11):1401–12. doi:10.1242/dmm.021147
38. Verner J, Trbusek M, Chovancova J, Jaskova Z, Moulis M, Folber F, et al. NOD/SCID IL2Rγ-null mouse xenograft model of human p53-mutated chronic lymphocytic leukemia and ATM-mutated mantle cell lymphoma using permanent cell lines. Leuk Lymphoma (2015) 56(11):3198–206. doi:10.3109/10428194.2015.1034701
39. Chen SS, Chiorazzi N. Murine genetically engineered and human xenograft models of chronic lymphocytic leukemia. Semin Hematol (2014) 51(3):188–205. doi:10.1053/j.seminhematol.2014.05.001
40. Ahrens AK, Chaturvedi NK, Nordgren TM, Dave BJ, Joshi SS. Establishment and characterization of therapy-resistant mantle cell lymphoma cell lines derived from different tissue sites. Leuk Lymphoma (2012) 53(11):2269–78. doi:10.3109/10428194.2012.691481
41. Hegde GV, Nordgren TM, Munger CM, Mittal AK, Bierman PJ, Weisenburger DD, et al. Novel therapy for therapy-resistant mantle cell lymphoma: multipronged approach with targeting of hedgehog signaling. Int J Cancer (2012) 131(12):2951–60. doi:10.1002/ijc.27602
42. Klanova M, Soukup T, Jaksa R, Molinsky J, Lateckova L, Maswabi BCL, et al. Mouse models of mantle cell lymphoma, complex changes in gene expression and phenotype of engrafted MCL cells: implications for preclinical research. Lab Invest (2014) 94:806–17. doi:10.1038/labinvest.2014.61
43. Tang JC, Ho FC, Chan AC, Chow EY, Srivastava G. Progression of spontaneous lymphomas in SJL mice: monitoring in vivo clonal evolution with molecular markers in sequential splenic samples. Lab Invest (1998) 78:1459–66.
44. Fredrickson TN, Lennert K, Chattopadhyay SK, Morse HC III, Hatley JW. Splenic marginal zone lymphomas of mice. Am J Pathol (1999) 154:805–12. doi:10.1016/S0002-9440(10)65327-8
45. Hough MR, Reis MD, Singaraja R, Bryce DM, Kamel-Reid S, Dardick I, et al. A model for spontaneous B-lineage lymphomas in IgHmu-HOX11 transgenic mice. Proc Natl Acad Sci U S A (1998) 95:13853–8. doi:10.1073/pnas.95.23.13853
46. Ward JM, Tadesse-Heath L, Perkins SN, Chattopadhyay SK, Hursting SD, Morse HC III. Splenic marginal zone B-cell and thymic T-cell lymphomas in p53-deficient mice. Lab Invest (1999) 50:3–14.
47. Drusco A, Pekarsky Y, Costinean S, Antenucci A, Conti L, Volinia S, et al. Common fragile site tumor suppressor genes and corresponding mouse models of cancer. J Biomed Biotechnol (2011) 4(6):1–10. doi:10.1155/2011/984505
48. Gilling CE, Mittal AK, Chaturvedi NK, Iqbal J, Aoun P, Bierman PJ, et al. Lymph node-induced immune tolerance in chronic lymphocytic leukaemia: a role for caveolin-1. Br J Haematol (2012) 158(2):216–31. doi:10.1111/j.1365-2141.2012.09148.x
49. Zou W, McDanela L, Smith LM. Caveloin-1 haploinsufficiency leads to partial transformation of human breast epithelial cells. Anticancer Res (2003) 23(6):4581–6.
50. Lee H, Park DS, Razani B, Russel RG, Pestell RG, Lisanti MP. Caveolin-1 mutations (P132L and null) behaves in a dominant-negative manner and caveolin01 (-/-) null mice show mammary epithelial cell hyperplasia. Am J Pathol (2002) 161(4):1357–69. doi:10.1016/S0002-9440(10)64412-4
51. Shukla A, Cutucache CE, Sutton GL, Pitner MA, Rai K, Rai S, et al. Absence of caveolin-1 leads to delayed development of chronic lymphocytic leukemia in Eµ-TCL1 mouse model. Exp Hematol (2016) 44(1):30.e–7.e. doi:10.1016/j.exphem.2015.09.005
52. Tomassian T, Humphries LA, Liu SD, Silva O, Brooks DG, Miceli MC. Caveolin-1 orchestrates TCR synaptic polarity, signal specificity, and function in CD8 T cells. J Immunol (2011) 187(6):2993–3002. doi:10.4049/jimmunol.1101447
Keywords: splenic marginal zone lymphoma, 7q minimally deleted region, 7q LOH, genetically engineered, neoplasms
Citation: Patten CL and Cutucache CE (2016) Murine Models of Splenic Marginal Zone Lymphoma: A Role for Cav1? Front. Oncol. 6:258. doi: 10.3389/fonc.2016.00258
Received: 30 July 2016; Accepted: 01 December 2016;
Published: 14 December 2016
Edited by:
Giuseppe Giaccone, Georgetown University, USAReviewed by:
Laura Gragnani, University of Florence, ItalyAlberto Arribas, Institute of Oncology Research (IOR), Switzerland
Copyright: © 2016 Patten and Cutucache. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine E. Cutucache, ccutucache@unomaha.edu