 Gillipsie Minhas
Gillipsie Minhas Jyoti Sharma
Jyoti Sharma Nooruddin Khan
Nooruddin Khan- Department of Biotechnology and Bioinformatics, School of Life Sciences, University of Hyderabad, Hyderabad, Telangana, India
Ischemia–reperfusion injury is a well-known pathological hallmark associated with diabetic retinopathy, glaucoma, and other related retinopathies that ultimately can lead to visual impairment and vision loss. Retinal ischemia pathogenesis involves a cascade of detrimental events that include energy failure, excitotoxic damage, calcium imbalance, oxidative stress, and eventually cell death. Retina for a long time has been known to be an immune privileged site; however, recent investigations reveal that retina, as well as the central nervous system, elicits immunological responses during various stress cues. Stress condition, such as reperfusion of blood supply post-ischemia results in the sequestration of different immune cells, inflammatory mediators including cytokines, chemokines, etc., to the ischemic region, which in turn facilitates induction of inflammatory conditions in these tissues. The immunological activation during injury or stress per se is beneficial for repair and maintenance of cellular homeostasis, but whether the associated inflammation is good or bad, during ischemia–reperfusion injury, hitherto remains to be explored. Keeping all these notions in mind, the current review tries to address the immune response and host stress response mechanisms involved in ischemia–reperfusion injury with the focus on the retina.
Introduction
Retinal ischemia is a condition that has been found to be connected to a large number of retinal diseases such as glaucoma, diabetic retinopathy, and central retinal artery occlusion, which are a leading cause of visual impairment or blindness (1–3). Ischemia, in general, is a condition that occurs due to disruption in blood supply to a particular tissue or organ, which cuts the supply of oxygen and glucose, triggering a cascade of events that ultimately ends with cell death. Retina being highly metabolic has very high oxygen consumption in the body (4). It is sensitive to oxygen deficiency, thus making it more susceptible to ischemic injury. Retina being an extension of central nervous system (CNS), it makes retina an ideal model system not only to examine the pathophysiology behind ischemia/hypoxia but also to assess different therapeutic strategies in animal models before proceeding to clinical trials, which can also be extrapolated to the brain (5, 6).
Ischemic cascade consists of energy failure, calcium influx followed by depolarization, and oxidative stress (7). Inflammation is an important phenomenon in the progression of any injury including ischemic injury (8). It usually helps in repair mechanism, but chronic inflammation causes more damage than good, triggering the release of reactive oxygen species (ROS), and tissue destruction (9). In ischemia–reperfusion injury, a similar condition is observed during the reperfusion, which is the restoration of blood supply to the ischemic tissue. The reoxygenation of tissue after ischemia causes more destruction by the production of ROS that damage the biomolecules by activation of inflammatory responses (10). In the current review, we have focused on the immune processes observed during ischemia–reperfusion injury, especially in the retina.
Retina: Immune Privileged Site
For a long time, retina and CNS have been considered as an immune privileged site due to its inability to post an immune response. The immune cells, which play the usual function of processing and presenting the antigens in the periphery, to our knowledge have not been reported in the retinal or CNS tissues; as a result, CNS is unable to mount an adaptive immune response (11). Investigations have also shown the presence of elevated levels of anti-inflammatory cytokines, which makes the natural environment in the brain as anti-inflammatory (12). Eye also has its similar mechanisms to control immune activation, and it contains many factors in aqueous humor that have shown to decrease the IFNγ production through the presence of anti-inflammatory factors (e.g., TGFβ2), which has demonstrated to reduce TCR activation (13–15).
The blood–brain barrier (BBB) provides anatomical and physiological protection to CNS. It restricts the migration of T lymphocytes and other immune mediators to brain through tight junctions between endothelial cells and helps in the maintenance of CNS as a unique immune privileged site (16, 17). On the similar lines, blood–retina barrier (BRB) is known to maintain homeostasis in the retina, which is essential for maintenance of immune privilege in the eye (18). The structure of BRB allows sustaining this condition in the eye. It is composed of two layers of tight junctions: the inner junction is present between retinal capillary endothelial cells and the outer one is between the RPE (18, 19). Many factors have been studied which affect the permeability of BRB, such as oxidative stress, VEGF, and inflammation (20, 21).
Recent studies have shown that the CNS tissues have specific immune responses against different kinds of trauma, infection, or injury, and the term “immune privilege” has become implicit (22). All the abovementioned factors and barriers make the CNS and retinal tissue isolated from the immune response; however, still, these components interact with the peripheral immune system (15, 23). Figure 1 enlists different aspects of cellular and immune response associated with retinal ischemia–reperfusion injury, which have been explored through this review.
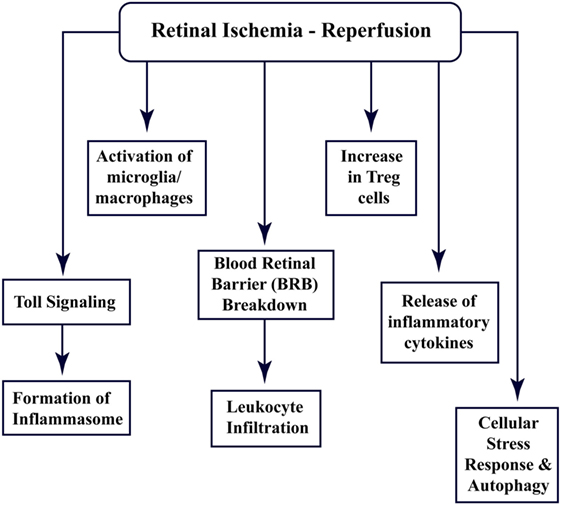
Figure 1. Multi-faceted cellular responses during retinal ischemia–reperfusion injury. Different aspects of immune signaling and host responses associated with ischemia–reperfusion injury in retina.
Ischemia–Reperfusion: Sterile Inflammation
Inflammation is a crucial component of host immune response essential for defense against invading pathogens, and it involves activation of different immune cells and the release of cytokines, chemokines, and other effector molecules. However, there are injuries which do not include any pathogen invasion but still invoke an inflammatory response, such as ischemia and trauma; these are identified as sterile inflammation (24).
In the case of the microbe-induced inflammation, it is activated through pattern-recognition receptors (PRRs), and these receptors recognize different pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), dsRNA, toxins, and other foreign molecules. In contrast, sterile inflammation involves damage-associated molecular patterns (DAMPs) (24). The DAMPs operate in the same way as the PAMPs but are endogenous instead of being pathogen derived. DAMPs are released by necrotic or apoptotic cells during injury and include proteins such as high-mobility group box-1 (HMGB1), mitochondrial components, uric acid, and others (25). Both PAMPs and DAMPs act through toll-like receptors (TLRs), NOD-like receptors (NLRs), and C-type lectin receptors, which are the common PRRs that are present on immune cells (26).
Toll-Like Receptor Signaling
Toll signaling is a pathway that is known to be activated in response to many diseases such as ischemia. This pathway was first identified in Drosophila, where it is essential for embryonic development (27–29). Subsequent studies demonstrated its presence in mammals (30, 31). TLRs are evolutionarily conserved membrane proteins with leucine-rich repeats and extracellular ligand-binding domains that recognize the PAMPs and DAMPs (32). TLR4 was the first component to be identified in mammals, which showed activation by bacterial components. These bacterial cell wall components upon binding to TLR activate the release of cytokines that assist in clearance of microbes, but in the case of excessive activation, it can become detrimental to host cells (33). Although it is now well established that TLR activation is associated with pathogen invasion, TLR has also been shown to be activated by DAMPs released by cells under stress. One such molecule is HMGB1, which is released by necrotic or apoptotic cells (34). Activation of TLR signaling, in turn, stimulates NFκB, a transcription factor that regulates the expression of genes responsible for cell adhesion, innate immune response, and inflammation (35, 36). Figure 2 shows the activation of TLR upon stimulation by DAMPs and downstream signaling pathways in response to ischemia–reperfusion, which through p38/MAPK signaling and NFκB leads to expression of inflammatory cytokines and activation of inflammasomes (37).
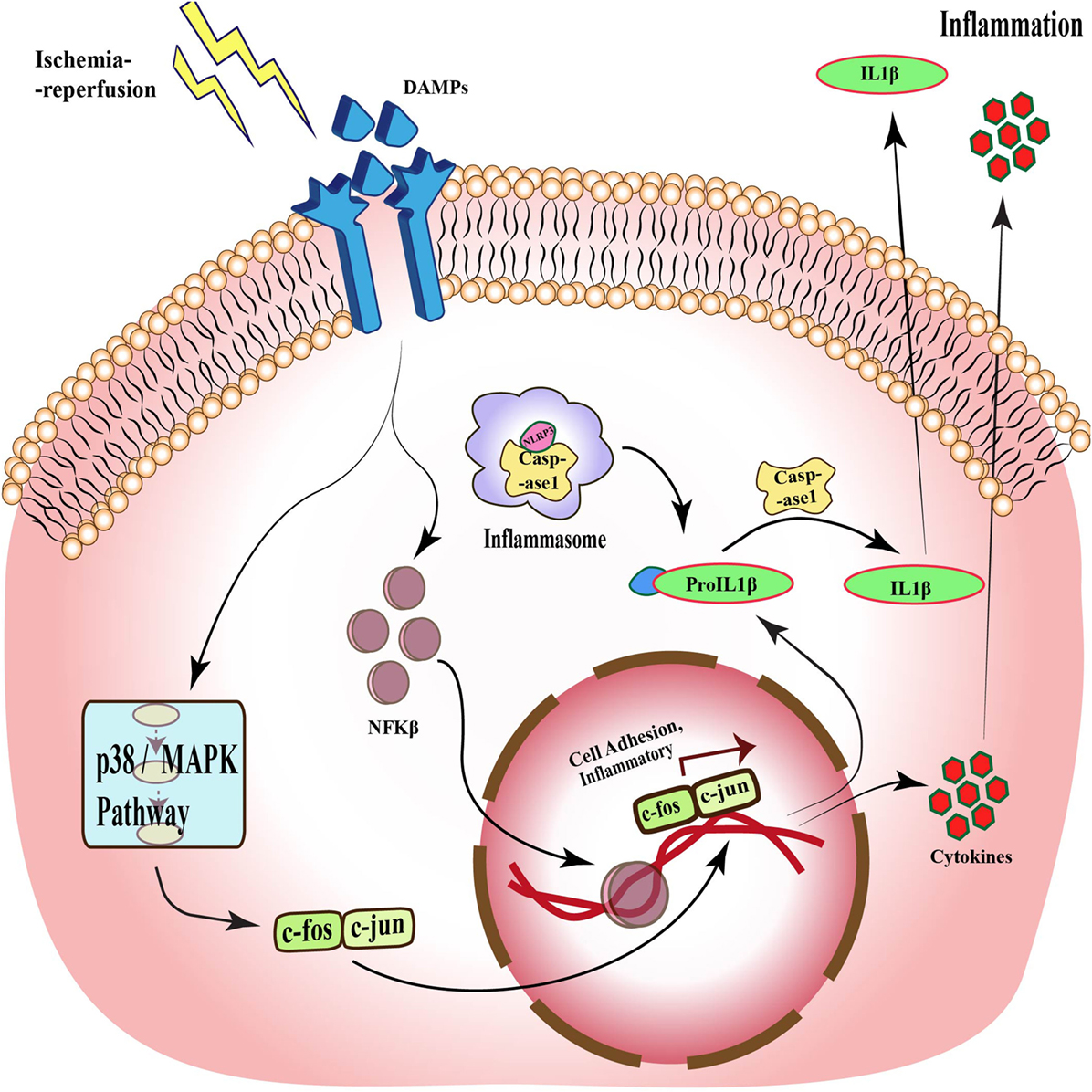
Figure 2. Toll-like receptor signaling during ischemia–reperfusion injury. Specific receptors, such as TLRs, are activated during ischemia–reperfusion through damage-associated molecular patterns (DAMPs), which activate downstream MAPK pathway and via different transcription factors such as NFκB and c-fos-c-jun, and translate different cell adhesion and inflammatory molecules, which causes inflammation. This pathway also acts through inflammasomes formation and through caspase 1 activation, which also results in inflammation.
Studies from different animal models have shown the important role of TLR signaling in ischemia–reperfusion injury (38–42). Hua et al. investigated TLR signaling in global cerebral ischemia and revealed neuronal death and increased expression of proinflammatory cytokines in wild-type mice subjected to ischemic injury. In the absence of TLR4, less infarct volumes with reduced cell death were observed in the cerebral ischemia mouse model, which demonstrates the deleterious role for TLR in ischemia–reperfusion injury (41). Lehnardt et al. showed similar outcome with TLR2-deficient mice, which developed less injury as compared to wild-type subjected to middle cerebral artery occlusion (MCAO) (43). More studies done to delineate the mechanism behind the protective role of TLR deletion have shown the contribution of PI3K/Akt pathway, which is already known to prevent apoptosis (44, 45). The outcomes observed in the brain were also reflected in retinal ischemia–reperfusion injury. Qi et al. generated retinal ischemia injury in rats by clamping retinal arteries and demonstrated the TLR4 activation post-injury (46). Studies have shown that TLR4-deficient mice are neuroprotective in ischemia–reperfusion (47). Kilic et al. exhibited less ischemic damage post-ischemia in focal cerebral ischemia as well as retinal ganglion cells (RGCs) degeneration in a TLR4 knock-out model (48). He et al. also demonstrated attenuation in neovascularization in retina along with less microglial activation and proinflammatory cytokines levels in TLR4-deficient mice subjected to ischemia (49). These studies emphasize the TLRs as prospective therapeutic targets to regulate ischemia–reperfusion injury.
Activation of Inflammasomes
Inflammasomes are the component of innate immune response, which have been implicated in different metabolic and neurodegenerative diseases. Inflammasomes are intracellular, multimeric protein complexes that are formed post-detection of PAMPs or DAMPs by specific PRRs. Inflammasome activation stimulates the expression of IL1β, IL18, and other proinflammatory cytokines downstream to initiate the cascade through caspase 1 (50, 51). Mechanistically, inflammasomes recruit pro-caspase 1, which further oligomerizes and auto-cleaves to form active caspase 1 that cleaves the pro-forms of cytokines IL1β and IL18 into their active forms (52).
The inflammasomes are generally named based on the scaffold protein that is associated with it. The most common being the NLRs, which also belong to the family of PRRs and are analogous to TLRs (53). The best-investigated inflammasomes that have been found to be associated with neuroinflammation are NACHT domain-LRR domain and pyrin domain containing protein (NALP) and nucleotide-binding oligomerization domain-like receptor with pyrin domain protein (NLRP1 and 3) (54, 55). The activation of NLRP inflammasomes through TLR4 activation has been demonstrated in a retinal ischemia–reperfusion injury model induced by ligation of retinal blood vessels in rat (46). Chi et al. also investigated the role of inflammasomes in RGC death due to retinal ischemia–reperfusion injury in a caspase 1-independent pathway. The study revealed that the ischemic injury increases the levels of TLR4, which further stimulates IL1β production through caspase 8 pathway (56). These revelations make the inflammasome activation a probable therapeutic for retinal ischemic injuries that will help to regulate inflammatory responses. It has been demonstrated in an Nlrp3 knock-out mice that the absence of inflammasomes delayed the progression of CNS injury (57).
Activation of Microglia–Macrophages
Microglia and astrocytes are the resident immune cells present in CNS. These are the first line of defense against any injury or infection in the CNS. The role of microglia and macrophages is controversial with different studies supporting the impact of their activation after the injury (58–60). Studies have shown that the activation of these cells can either be beneficial or detrimental for a tissue. As is seen in the case of Alzheimer’s, where the activation of microglia helps in clearance of amyloid load and hence is beneficial (61, 62). Similarly, activated macrophages assist in removal of leukocytes through phagocytosis (63). In retina, Müller glia are the principal glial cells. Müller glia are the radial glia, which are found throughout the thickness of the retina and are associated with a majority of retinal degenerations. In normal physiology, Müller glia maintain the layer arrangement in the retina, provide trophic support, and remove the waste, whereas under the pathological conditions, these cells undergo reactive gliosis. The involvement of Müller glia in ischemic injury has been implicated through different animal models, and it has been shown to play protective as well as destructive role in retinal degenerations (64–67). Wurm et al. investigated the impact of ischemia–reperfusion injury on the Müller glial cells in porcine retina exposed to high intraocular pressure (IOP). The authors demonstrated swelling and gliosis through increased expression of glial fibrillary acidic protein (GFAP) and vimentin in Müller glia (68).
Rangasamy et al. have shown the activation of microglia and macrophages in an in vivo as well as the in vitro model of diabetic retinopathy, another ischemia-associated condition (69). In in vitro, the authors exposed the retinal endothelial cells to high glucose conditions and demonstrated an increase in expression levels of macrophage marker and chemokine ligand (CCl2). A similar response was observed in the rat model of diabetes with elevated macrophage and microglia in the retina (69). The activated glia after ischemic injury releases cytokines, such as VEGF, which assist in angiogenesis and resupply of blood (70). Microglia can also produce neurotrophic factors for cell survival. On the contrary, microglia can also release TNFα and IL1β and aggravate the neuronal damage (71, 72). The role of glial cells in the ischemic injury still needs additional investigations to target them as potential therapeutics.
Infiltration of Leukocytes: Breach in Blood–Retina Barrier
Ischemia–reperfusion injury also has an impact on microcirculation in the tissue, which leads to infiltration of inflammatory cells due to increased permeability (73). The inner retinal barrier is known to be disrupted in many retinal diseases including ischemia (20, 74). Wilson et al. visualized leakage in BRB in a rabbit model of IOP-induced ischemia–reperfusion injury through magnetic-resonance imaging (75). The ischemia–reperfusion injury also impacts the permeability by affecting the different tight-junction proteins that constitute the BRB, such as occludin and zona occludens. Muthusamy et al. in their study in IOP-induced retinal ischemia model in rats have investigated the tight-junction proteins after injury and have shown decreased levels of occludin, which results in the increased vascular permeability and hence infiltration of leukocytes (76, 77). Tsujikawa in their study on rats subjected to optic nerve ligation revealed leukocyte–endothelium interaction through fluorescein angiography (78). Cell adhesion molecules, such as selectins, ICAM1, integrins, and CD11/CD18 have also shown to play a significant role in leukocyte trafficking and infiltration in ischemia (79). Stoll et al. observed an increase in the levels of ICAM and selectin after focal cerebral ischemia, which led to permeation of T cells and macrophages (80). Retinal inflammation has been shown to involve adhesion of leukocytes to the retinal blood vessels, thus affecting the integrity of BRB (69). These molecules can also be exploited as therapy through a decrease in trafficking of immune cells. Tsujikawa et al. in their another study blocked adhesion molecules using specific antibodies, resulting in less leukocyte accumulation during ischemia–reperfusion injury (81). As already discussed in previous sections, reperfusion of blood not only increases oxygen and glucose but also exacerbates the ischemic injury by activating inflammatory responses and promoting immune cells infiltration (9, 82). Lymphocytes, a sub-population of leukocytes, have also been associated with ischemic injury (83). Studies have demonstrated accumulation of T-cells (CD4+ and CD8+ cells) at the site of ischemic injury in the brain, where different knock-out mice were used to investigate the role of T lymphocytes in cerebral ischemia (84). T lymphocytes have shown their presence at as early as 24 h after ischemic injury, which peaked at day 3 after the injury (85). When cerebral ischemic injury was induced in SCID or Rag−/− mice, which lack the T- and B-cells, less damage and smaller infarct volumes were observed (84). Involvement of T-cells, especially the IL17-producing cells, has also already been demonstrated in different studies in the brain (86, 87). In cerebral ischemia, it has been observed that the γδT cells, a distinct T cell type, are the major IL17-producing cells (88), and the activation of these cells is the primary response to ischemia, which further activates matrix metalloproteinase, pro-inflammatory cytokines, and chemokines that are involved in aggravation of the ischemia–reperfusion injury. A decrease in infarct volume has been demonstrated in TCR-γδ knock-out mice subjected to ischemia (89). Shichita et al. in their study also highlighted a central role of γδT cells in the pathology of cerebral ischemia (90). These studies indicate that targeting T cells could be a novel therapeutic strategy for constraining inflammation during ischemia–reperfusion injury. Apart from these studies, investigations are also required to elucidate the role of T cells-mediated immune modulation during retinal ischemia. Figure 3 gives a brief overview of leukocyte trafficking due to breach in blood–retina barrier after ischemic injury to retina, along with the proposed mechanism about how different subtypes of T cells are involved in ischemia, such as CD4+ T regulatory cells that release anti-inflammatory TGFβ, IL10 (91), and γδT cells that produce IL17 through IL23 stimulation (92), along with the production of IL8 and VEGF from different T-cell subtypes (93).
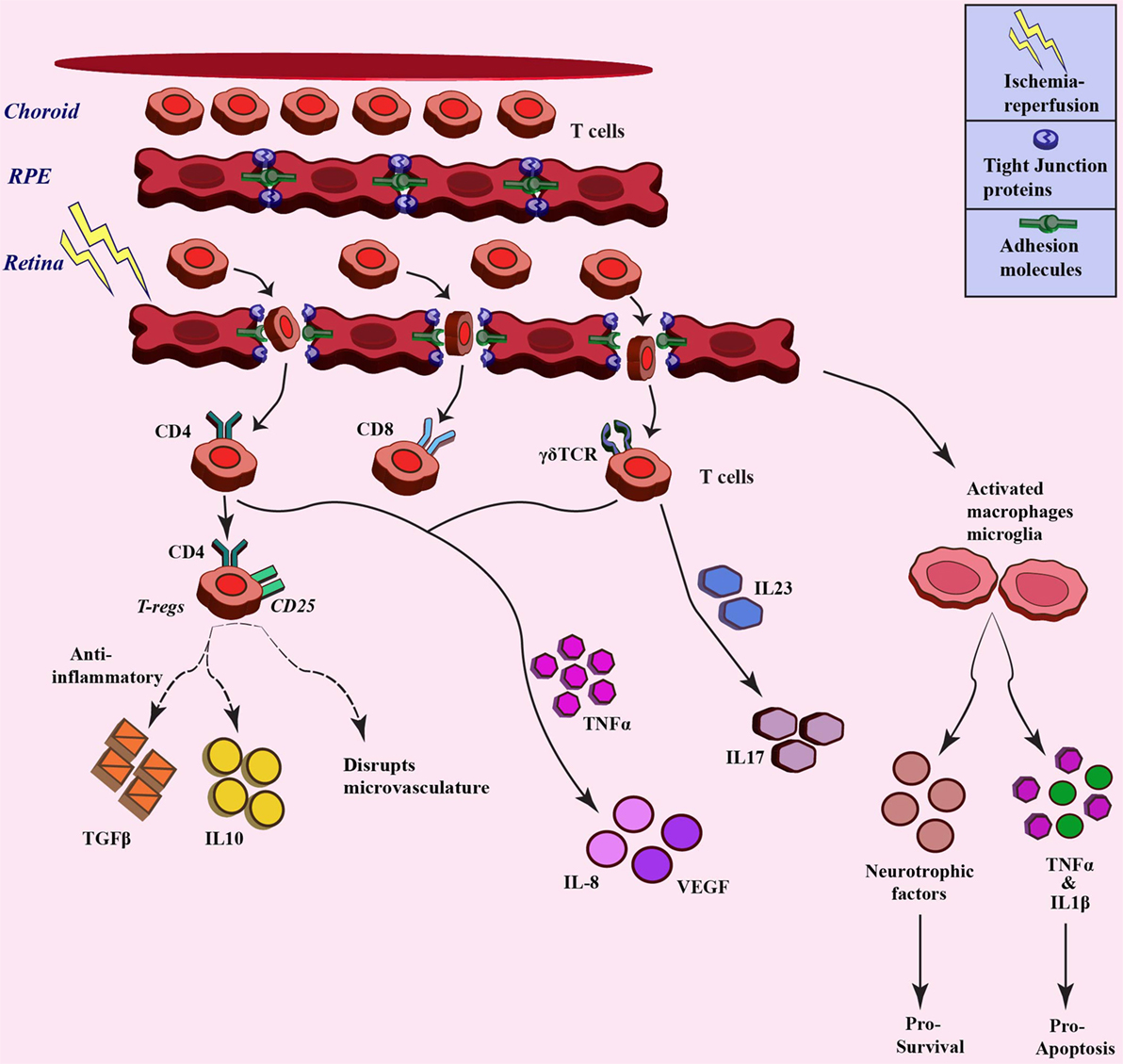
Figure 3. Trafficking of T cells after ischemia–reperfusion and related downstream response. Different T cell populations involved in ischemia–reperfusion injury pathogenesis. These T cells infiltrate upon breach of blood-retina barrier due to ischemia injury and different populations stimulate varied downstream signaling through cytokine/chemokine mediators (broken lines denotes the paradox outcomes).
Protector or Promoter: Regulatory T Cells
Balance of immune responses is essential not only to clear the pathogens but also to control unwanted immune response. Regulatory T cells (Tregs) are the cells which were first identified in the mid-1990s as the cells that control the immune responses through feedback mechanisms (94, 95). These cells also protect the host against any self-antigens and any related auto-immune disease; however, later, it was identified that these cells also have a role to play in the suppression of infections, including uveoretinitis (96). The CD4+ T cells principally can be divided into two sub-populations, Th cells that activate immunity and Treg cells that keep a check on the Th cells activity. Treg cells are more precisely identified as the CD4+ CD25+ population and are characterized by the presence of transcription factor, fork-head box protein 3 (Foxp3) (97, 98). In the case of ischemia, there have been studies with paradox outcomes. Liesz et al. demonstrated a neuroprotective role of Tregs in stroke, with less infarct volumes accompanied by a decrease in levels of proinflammatory cytokines. The authors depleted the Treg cell population and observed an increase in secondary damage to brain post-ischemia along with a higher number of infiltrating leukocytes and activated microglia (99). Treg cells also secrete anti-inflammatory cytokines, such as IL10 and TGFβ. On the contrary, Stubbe et al. in their study on brain subjected to MCAO-induced ischemia reported the detrimental role of Treg cells (100). Kleinschnitz demonstrated that Tregs promote the ischemic injury and impact the microvasculature in the brain. The authors tested the effect of injury in depletion of regulatory T cells (DEREG) mice, which are devoid of any Tregs and revealed less infarct volumes after MCAO (101). Consequently, Treg cells, on the one hand, can protect the brain from ischemic injury by modulating inflammation and increasing the expression of metallomatrix proteases; on the other hand, these cells can also aggravate the ischemic insult by disturbing the microvasculature and hindering functional recovery (102).
Retina-specific Treg cells are produced in response to antigens found in the retina. These can either be natural Tregs, which are generated against retinal antigens expressed in the thymus, or these can be generated independently of thymus from mature peripheral T cells based on exposure to antigens (103). In the retina, this cell population has been associated with uveoretinitis, where an increased number of CD4+CD25+ Tregs was detected in spleen and eye (104). More studies are required to investigate the role of Tregs in the retina and to translate the findings from cerebral ischemia to the retinal ischemia.
Molecular Trigger: Inflammatory Cytokines
Cytokines are the molecules released by immune cells after an injury, which can be either anti-inflammatory or proinflammatory in nature. Ischemic injury too is associated with inflammation and release of cytokines. The cytokines induce the migration of leukocytes in the ischemic tissue, which in turn release more cytokines and exacerbate the ischemic injury (105). Both Th1- and Th2-based immune response have been linked with specific proinflammatory cytokine signaling observed in retinal damage. Tumor necrosis factor α (TNF-α) is a proinflammatory cytokine, produced by microglia and leukocytes as an early response toward ischemia–reperfusion. The levels of TNFα were found to be upregulated in porcine retina 5–12 h after ischemia–reperfusion (106). In another model of retinal ischemia produced by an increase in IOP, upregulation in levels of TNF-α as well as its receptors, TNFR1 and TNFR2, was noted as early as 6 h after the reperfusion (107). The increase in TNF-α levels has also been reported by Yoshida et al. in ischemic retina. The expression was found to be localized in macrophages and microglia (108). TNF-α expression further increases the levels of other cytokines, such as IL-8 and VEGF (108, 109). IL17 and IFNγ mRNA levels were also found to be elevated in brain tissue and peripheral blood of permanent MCAO rat models (110). IL17 production has also been identified in human stroke patients (111). IL23 knock-out mice demonstrated smaller infarct volumes (90). IL23 mediates production of IL17 through γδT cells (112). IL6, another Th2 response-related proinflammatory cytokine was detected after reperfusion in high IOP-induced retinal injury in rats (113). Hangai et al. demonstrated many fold increase in IL1α and β levels up to 12 h after ischemia induced by optic nerve ligation in rats (114). These inflammatory cytokines can also be targeted as therapy for ischemia–reperfusion injury. Berger et al. demonstrated improvement in retinal function on treatment with TNFα antibody in high IOP rat model of ischemia–reperfusion (115). Studies have demonstrated protective role of anti-inflammatory interventions in ischemic injury models induced by high IOP as well as related retinal degenerations, such as diabetic retinopathy in rats (116–118).
Cellular Stress Responses and Immune Regulations During Ischemia
The cellular stress response is the homeostatic mechanism, which enables cells to adapt to various stress cues such as ischemia. Cell senses varieties of stress conditions via different sensors such as general control non-derepressible 2 (GCN2), which is activated during amino acid deprivation, heme-regulated inhibitor kinase (HRI), senses heme deficiency, protein kinase RNA (PKR)-like endoplasmic reticulum (ER) kinase (PERK), and gets activated during viral infections. During stress condition, these sensors get phosphorylated, which further phosphorylate the eukaryotic initiation factor (eIF2α), resulting in the attenuation of active polysome formation and global protein synthesis. The untranslated mRNA recruits different RNA-binding proteins (RBPs), which together form a structure known as “riboclusters.” These clusters dictate the fate of the mRNA transcripts (119, 120). Riboclusters formed under stress environment are also known to regulate the balance between inflammatory and anti-inflammatory cytokines through different pathways (119). ER is the primary organelle that is responsible for synthesis, folding, and trafficking of proteins. ER also participates in the detection of any metabolic changes to the cell through different cellular sensors, such as PERK, IRE1, and ATF6. Any disturbance in the normal physiology can lead to ER stress and hence activate these cellular sensors and associated stress response pathways in the form of unfolded protein response (UPR). These stress detectors are inactive under normal condition by glucose-regulated protein (GRP78), also known as BiP. Excessive stress removes the GRP78 from the sensors and makes them available for downstream response pathway. The UPR pathway responds to this stress by halting the protein translation and removing the misfolded proteins that have accumulated through chaperones. Prolonged stress can result in inflammation and related damage and ultimately, apoptosis, if the cell homeostasis is not regained (121, 122). ER stress has been implicated in various neurodegenerative and vascular diseases including different retinal degeneration diseases, Stargardt’s, age-related macular degeneration, and retinitis pigmentosa (123). Doh et al. have related ER stress with glaucoma, where high IOP stimulated the expression of BiP, CHOP, and phosphor-PERK along with ganglion cell death (124).
Prolonged stress including nutrition deprivation could also result in activation of homeostatic processes such as autophagy (125). Autophagy is a conserved defense mechanism that regulates protein turn-over as a normal phenomenon but is found to be upregulated in response to stress. Autophagy plays a crucial role in regulating immune responses including T-cell response. Coronary artery occlusion has shown to result in the increased levels of LC3-II conversion, a well-established marker for autophagy (126, 127). Different studies have investigated the role of autophagy in animal models of ischemic injury in the retina. Piras et al. demonstrated autophagy 24 h after in a retinal ischemia model induced by increasing the IOP (128). A similar outcome has been observed by Wei et al. with the accumulation of autophagosomes and high levels of LC3-II in RGCs, 6 h after the injury (129). The knowledge about cellular stress sensors and response pathways pertaining to retinal degeneration is still at an initial stage, which demands further investigations to bring out target therapies.
Summary
Ischemia-induced retinal injury is associated with many diseases such as diabetic retinopathy and glaucoma, which result in vision impairment and blindness. With no available cure so far for retinal ischemia, it has become pertinent to identify potential targets for designing therapies. Immune modulation is one aspect that could be a potential therapeutic target for the ischemic injury. Furthermore, a better understanding about cellular stress responses can also help in future, to design better interventions for retinal ischemia–reperfusion injuries. All together, it has to be kept in consideration that since ischemia is a consequence of synergistic effects of different cell types and mechanisms; therefore, targeting a single cell or molecule will not reveal any progressive therapeutic strategy. Moreover, since ischemia is a dynamic injury, it is crucial to investigate time-dependent inflammatory responses involved so as to keep the timing for therapy in consideration with minimal damage.
Author Contributions
GM wrote the paper; JS made the figures; and NK contributed to writing, editing, and designing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work has been supported by the Department of Biotechnology, Government of India (BT/PR8624/MED/29/798/2013), the Nano Mission Council, Department of Science and Technology (DST No: SB/YS/LS-163/2013), and the University Grants Commission, Government of India (MRP-MAJOR-BIOT-2013-40689).
Abbreviations
ATF6, activating transcription factor 6; BBB, blood–brain barrier; BRB, blood–retina barrier; CCL2, C–C motif chemokine ligand 2; CD, cluster of differentiation; CHOP, CCAAT-enhancer-binding protein homologous protein; CNS, central nervous system; DAMP, damage-associated molecular pattern; DEREG, depletion of regulatory T cell; eIF2α, eukaryotic initiation factor; ER, endoplasmic reticulum; GCN2, general control non-derepressible 2; GFAP, glial fibrillary acidic protein; GRP78, 78 kDa glucose-regulated protein; HMGB1, high-mobility group box 1; HRI, heme-regulated inhibitor kinase; ICAM-1, intracellular adhesion molecule 1; IFNγ, interferon gamma; IL1β, interleukin 1beta; IL18, interleukin 18; IOP, intraocular pressure; IRE1, inositol-requiring enzyme 1; ISR, integrated stress response; LC3, microtubule-associated protein light chain 3; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinases; MCAO, middle cerebral artery occlusion; mRNA, messenger ribonucleic acid; NLR, NOD-like receptor; NOD, nucleotide oligomerization domain; PAMP, pathogen-associated molecular pattern; PERK, protein kinase RNA-like endoplasmic reticulum kinase; PI3K, phosphatidylinositol-3-kinase; PKR, protein kinase RNA-activated; PRR, pattern-recognition receptor; RBPs, RNA-binding proteins; RGCs, retinal ganglion cells; ROS, reactive oxygen species; RPE, retinal pigment epithelium; SCID, severe combined immunodeficiency; TCR, T-cell receptor; TGFβ2, transforming growth factor 2; TLR, toll-like receptor; TNF-α, tumor necrosis factor-alpha; TNFR, tumor necrosis factor receptor; UPR, unfolded protein response; VEGF, vascular endothelial growth factor.
References
1. Osborne NN, Casson RJ, Wood JPM, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res (2004) 23(1):91–147. doi:10.1016/j.preteyeres.2003.12.001
2. Osborne NN, Ugarte M, Chao M, Chidlow G, Bae JH, Wood JPM, et al. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv Ophthalmol (1999) 43(Suppl 1):S102–28. doi:10.1016/S0039-6257(99)00044-2
3. Bresnick GH, De Venecia G, Myers FL, Harris JA, Davis MD. Retinal ischemia in diabetic retinopathy. Arch Ophthalmol (1975) 93(12):1300–10. doi:10.1001/archopht.1975.01010020934002
4. Ames A III. Energy requirements of CNS cells as related to their function and to their vulnerability to ischemia: a commentary based on studies on retina. Can J Physiol Pharmacol (1992) 70(S1):S158–64. doi:10.1139/y92-257
5. Ames A, Nesbett F. In vitro retina as an experimental model of the central nervous system. J Neurochem (1981) 37(4):867–77. doi:10.1111/j.1471-4159.1981.tb04473.x
6. London A, Benhar I, Schwartz M. The retina as a window to the brain – from eye research to CNS disorders. Nat Rev Neurol (2013) 9(1):44–53. doi:10.1038/nrneurol.2012.227
7. Kaplan J, Dimlich RV, Biros MH, Hedges J. Mechanisms of ischemic cerebral injury. Resuscitation (1987) 15(3):149–69. doi:10.1016/0300-9572(87)90012-8
8. del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol (2000) 10(1):95–112. doi:10.1111/j.1750-3639.2000.tb00247.x
9. Jean WC, Spellman SR, Nussbaum ES, Low WC. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery (1998) 43(6):1382–96;discussion1396–7. doi:10.1097/00006123-199812000-00076
10. Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M. Molecular mechanisms of ischemia–reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol (2013) 47(1):9–23. doi:10.1007/s12035-012-8344-z
11. Engelhardt B, Coisne C. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS (2011) 8(1):1. doi:10.1186/2045-8118-8-4
12. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest (2012) 122(4):1164–71. doi:10.1172/JCI58644
14. Yoshida M, Kezuka T, Streilein JW. Participation of pigment epithelium of iris and ciliary body in ocular immune privilege. 2. Generation of TGF-β–producing regulatory T cells. Invest Ophthalmol Vis Sci (2000) 41(12):3862–70.
15. Cousins SW, Trattler WB, Streilein JW. Immune privilege and suppression of immunogenic inflammation in the anterior chamber of the eye. Curr Eye Res (1991) 10(4):287–97. doi:10.3109/02713689108996334
16. Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, Del Zoppo GJ, Doolittle ND, et al. Immunologic privilege in the central nervous system and the blood–brain barrier. J Cereb Blood Flow Metab (2013) 33(1):13–21. doi:10.1038/jcbfm.2012.153
17. Pachter JS, de Vries HE, Fabry Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol (2003) 62(6):593–604. doi:10.1093/jnen/62.6.593
18. Cunha-Vaz J. The blood-retinal barriers. Doc Ophthalmol (1976) 41(2):287–327. doi:10.1007/BF00146764
19. Cunha-Vaz J. The blood-ocular barriers. Surv Ophthalmol (1979) 23(5):279–96. doi:10.1016/0039-6257(79)90158-9
20. Kaur C, Foulds W, Ling E. Blood–retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog Retin Eye Res (2008) 27(6):622–47. doi:10.1016/j.preteyeres.2008.09.003
21. Kaur C, Ling E. Blood brain barrier in hypoxic-ischemic conditions. Curr Neurovasc Res (2008) 5(1):71–81. doi:10.2174/156720208783565645
22. Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun (2003) 17(1):13–9. doi:10.1016/S0889-1591(02)00055-7
23. Stein-Streilein J, Taylor AW. An eye’s view of T regulatory cells. J Leukoc Biol (2007) 81(3):593–8. doi:10.1189/jlb.0606383
24. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10(12):826–37. doi:10.1038/nri2873
25. Shen H, Kreisel D, Goldstein DR. Processes of sterile inflammation. J Immunol (2013) 191(6):2857–63. doi:10.4049/jimmunol.1301539
26. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi:10.1016/j.cell.2010.01.022
27. Belvin MP, Anderson KV. A conserved signaling pathway: the Drosophila toll-dorsal pathway. Annu Rev Cell Dev Biol (1996) 12(1):393–416. doi:10.1146/annurev.cellbio.12.1.393
28. Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature (1980) 287(5785):795–801. doi:10.1038/287795a0
29. Rosetto M, Engstrom Y, Baldari CT, Telford JL, Hultmark D. Signals from the IL-1 receptor homolog, toll, can activate an immune response in a Drosophila hemocyte cell line. Biochem Biophys Res Commun (1995) 209(1):111–6. doi:10.1006/bbrc.1995.1477
30. Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature (1997) 388(6640):394–7. doi:10.1038/41131
31. Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila toll. Proc Natl Acad Sci U S A (1998) 95(2):588–93. doi:10.1073/pnas.95.2.588
32. Beutler B. Inferences, questions and possibilities in toll-like receptor signalling. Nature (2004) 430(6996):257–63. doi:10.1038/nature02761
33. Anderson KV. Toll signaling pathways in the innate immune response. Curr Opin Immunol (2000) 12(1):13–9. doi:10.1016/S0952-7915(99)00045-X
34. Schlueter C, Weber H, Meyer B, Rogalla P, Röser K, Hauke S, et al. Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol (2005) 166(4):1259–63. doi:10.1016/S0002-9440(10)62344-9
35. Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-β in toll-like receptor-stimulated dendritic cell activation. Int Immunol (2002) 14(10):1225–31. doi:10.1093/intimm/dxf089
36. Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-κB is activated and promotes cell death in focal cerebral ischemia. Nat Med (1999) 5(5):554–9. doi:10.1038/8432
37. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol (2014) 5:461. doi:10.3389/fimmu.2014.00461
38. Arslan F, Smeets MB, O’Neill LA, Keogh B, McGuirk P, Timmers L, et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti–toll-like receptor-2 antibody. Circulation (2010) 121(1):80–90. doi:10.1161/CIRCULATIONAHA.109.880187
39. Arumugam TV, Okun E, Tang S-C, Thundyil J, Taylor SM, Woodruff TM. Toll-like receptors in ischemia-reperfusion injury. Shock (2009) 32(1):4–16. doi:10.1097/SHK.0b013e318193e333
40. Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL, et al. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg (2004) 128(2):170–9. doi:10.1016/j.jtcvs.2003.11.036
41. Hua F, Ma J, Ha T, Xia Y, Kelley J, Williams DL, et al. Activation of toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol (2007) 190(1):101–11. doi:10.1016/j.jneuroim.2007.08.014
42. Shen XD, Ke B, Zhai Y, Gao F, Busuttil RW, Cheng G, et al. Toll-like receptor and heme oxygenase-1 signaling in hepatic ischemia/reperfusion injury. Am J Transplant (2005) 5(8):1793–800. doi:10.1111/j.1600-6143.2005.00932.x
43. Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, et al. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol (2007) 190(1–2):28–33. doi:10.1016/j.jneuroim.2007.07.023
44. Cao C-X, Yang Q-W, Lv F-L, Cui J, Fu H-B, Wang J-Z. Reduced cerebral ischemia-reperfusion injury in toll-like receptor 4 deficient mice. Biochem Biophys Res Commun (2007) 353(2):509–14. doi:10.1016/j.bbrc.2006.12.057
45. Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation (2007) 115(12):1599–608. doi:10.1161/CIRCULATIONAHA.106.603431
46. Qi Y, Zhao M, Bai Y, Huang L, Yu W, Bian Z, et al. Retinal ischemia/reperfusion injury is mediated by toll-like receptor 4 activation of NLRP3 inflammasomesTLR4 regulates NLRP3 activation in retinal IR. Invest Ophthalmol Vis Sci (2014) 55(9):5466–75. doi:10.1167/iovs.14-14380
47. Dvoriantchikova G, Barakat DJ, Hernandez E, Shestopalov VI, Ivanov D. Toll-like receptor 4 contributes to retinal ischemia/reperfusion injury. Mol Vis (2010) 16:1907–12.
48. Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM. TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiol Dis (2008) 31(1):33–40. doi:10.1016/j.nbd.2008.03.002
49. He C, Sun Y, Ren X, Lin Q, Hu X, Huang X, et al. Angiogenesis mediated by toll-like receptor 4 in ischemic neural tissue. Arterioscler Thromb Vasc Biol (2013) 33(2):330–8. doi:10.1161/ATVBAHA.112.300679
50. Chakraborty S, Kaushik DK, Gupta M, Basu A. Inflammasome signaling at the heart of central nervous system pathology. J Neurosci Res (2010) 88(8):1615–31. doi:10.1002/jnr.22343
51. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol (2008) 9(8):857–65. doi:10.1038/ni.1636
52. Guo H, Callaway JB, Ting JPY. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21(7):677–87. doi:10.1038/nm.3893
53. Trendelenburg G. Molecular regulation of cell fate in cerebral ischemia: role of the inflammasome and connected pathways. J Cereb Blood Flow Metab (2014) 34(12):1857–67. doi:10.1038/jcbfm.2014.159
54. Martinon F, Gaide O, Pétrilli V, Mayor A, Tschopp J. NALP inflammasomes: a central role in innate immunity. Semin Immunopathol (2007) 29(3):213–29. doi:10.1007/s00281-007-0079-y
55. Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ (2007) 14(1):10–22. doi:10.1038/sj.cdd.4402038
56. Chi W, Li F, Chen H, Wang Y, Zhu Y, Yang X, et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1β production in acute glaucoma. Proc Natl Acad Sci U S A (2014) 111(30):11181–6. doi:10.1073/pnas.1402819111
57. Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A (2012) 109(26):10480–5. doi:10.1073/pnas.1201836109
58. Graeber MB. Changing face of microglia. Science (2010) 330(6005):783–8. doi:10.1126/science.1190929
59. Neumann H, Kotter MR, Franklin RJM. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain (2009) 132(2):288–95. doi:10.1093/brain/awn109
60. Stollg G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol (1999) 58(3):233–47. doi:10.1016/S0301-0082(98)00083-5
61. Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, et al. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol (2010) 177(5):2549–62. doi:10.2353/ajpath.2010.100265
62. Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J Neurosci (2010) 30(50):17091–101. doi:10.1523/JNEUROSCI.4403-10.2010
63. Meszaros AJ, Reichner JS, Albina JE. Macrophage-induced neutrophil apoptosis. J Immunol (2000) 165(1):435–41. doi:10.4049/jimmunol.165.1.435
64. Pannicke T, Uckermann O, Iandiev I, Biedermann B, Wiedemann P, Perlman I, et al. Altered membrane physiology in Müller glial cells after transient ischemia of the rat retina. Glia (2005) 50(1):1–11. doi:10.1002/glia.20151
65. Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, et al. Muller cells in the healthy and diseased retina. Prog Retin Eye Res (2006) 25(4):397–424. doi:10.1016/j.preteyeres.2006.05.003
66. Bringmann A, Reichenbach A. Role of Muller cells in retinal degenerations. Front Biosci (2001) 6:E72–92. doi:10.2741/Bringman
67. Bringmann A, Wiedemann P. Muller glial cells in retinal disease. Ophthalmologica (2012) 227(1):1–19. doi:10.1159/000328979
68. Wurm A, Iandiev I, Uhlmann S, Wiedemann P, Reichenbach A, Bringmann A, et al. Effects of ischemia–reperfusion on physiological properties of Müller glial cells in the porcine retina. Invest Ophthalmol Vis Sci (2011) 52(6):3360–7. doi:10.1167/iovs.10-6901
69. Rangasamy S, McGuire PG, Nitta CF, Monickaraj F, Oruganti SR, Das A. Chemokine mediated monocyte trafficking into the retina: role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS One (2014) 9(10):e108508. doi:10.1371/journal.pone.0108508
70. Nedergaard M, Dirnagl U. Role of glial cells in cerebral ischemia. Glia (2005) 50(4):281–6. doi:10.1002/glia.20205
71. Barone F, Arvin B, White R, Miller A, Webb C, Willette R, et al. Tumor necrosis factor-α A mediator of focal ischemic brain injury. Stroke (1997) 28(6):1233–44. doi:10.1161/01.STR.28.6.1233
72. Gregersen R, Lambertsen K, Finsen B. Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab (2000) 20(1):53–65. doi:10.1097/00004647-200001000-00009
73. Eltzschig HK, Collard CD. Vascular ischaemia and reperfusion injury. Br Med Bull (2004) 70(1):71–86. doi:10.1093/bmb/ldh025
74. Kaur C, Sivakumar V, Yong Z, Lu J, Foulds W, Ling E. Blood–retinal barrier disruption and ultrastructural changes in the hypoxic retina in adult rats: the beneficial effect of melatonin administration. J Pathol (2007) 212(4):429–39. doi:10.1002/path.2195
75. Wilson CA, Berkowitz BA, Funatsu H, Metrikin DC, Harrison DW, Lam MK, et al. Blood-retinal barrier breakdown following experimental retinal ischemia and reperfusion. Exp Eye Res (1995) 61(5):547–57. doi:10.1016/S0014-4835(05)80048-X
76. Muthusamy A, Lin C-M, Lindner H, Shanmugam S, Abcouwer SF, Antonetti DA. Molecular mechanisms of blood-retinal barrier breakdown in ischemia-reperfusion injury. Invest Ophthalmol Vis Sci (2012) 53(14):5327.
77. Muthusamy A, Lin C-M, Shanmugam S, Lindner HM, Abcouwer SF, Antonetti DA. Ischemia–reperfusion injury induces occludin phosphorylation/ubiquitination and retinal vascular permeability in a VEGFR-2-dependent manner. J Cereb Blood Flow Metab (2014) 34(3):522–31. doi:10.1038/jcbfm.2013.230
78. Tsujikawa A, Ogura Y, Hiroshiba N, Miyamoto K, Kiryu J, Honda Y. In vivo evaluation of leukocyte dynamics in retinal ischemia reperfusion injury. Invest Ophthalmol Vis Sci (1998) 39(5):793–800.
79. Seekamp A, Till G, Mulligan M, Paulson J, Anderson D, Miyasaka M, et al. Role of selectins in local and remote tissue injury following ischemia and reperfusion. Am J Pathol (1994) 144(3):592.
80. Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol (1998) 56(2):149–71. doi:10.1016/S0301-0082(98)00034-3
81. Tsujikawa A, Ogura Y, Hiroshiba N, Miyamoto K, Kiryu J, Tojo SJ, et al. Retinal ischemia-reperfusion injury attenuated by blocking of adhesion molecules of vascular endothelium. Invest Ophthalmol Vis Sci (1999) 40(6):1183–90.
82. McCord J. Oxygen-derived radicals: a link between reperfusion injury and inflammation. Fed Proc (1987) 46(7):2402–6.
83. Ysebaert DK, De Greef KE, De Beuf A, Van Rompay AR, Vercauteren S, Persy VP, et al. T cells as mediators in renal ischemia/reperfusion injury. Kidney Int (2004) 66(2):491–6. doi:10.1111/j.1523-1755.2004.761_4.x
84. Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-γ in ischemic stroke. Circulation (2006) 113(17):2105–12. doi:10.1161/CIRCULATIONAHA.105.593046
85. Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol (1994) 55(2):195–203. doi:10.1016/0165-5728(94)90010-8
86. Gelderblom M, Arunachalam P, Magnus T. γδ T cells as early sensors of tissue damage and mediators of secondary neurodegeneration. Front Cell Neurosci (2014) 8(368):10.3389. doi:10.3389/fncel.2014.00368
87. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood (2012) 120(18):3793–802. doi:10.1182/blood-2012-02-412726
88. Holtmeier W, Kabelitz D. gammadelta T cells link innate and adaptive immune responses. Chem Immunol Allergy (2005) 86:151–83. doi:10.1159/000086659
89. Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vδ1+ γδ T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol (2007) 178(7):4466–72. doi:10.4049/jimmunol.178.7.4466
90. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing γδT cells in the delayed phase of ischemic brain injury. Nat Med (2009) 15(8):946–50. doi:10.1038/nm.1999
91. Corthay A. How do regulatory T cells work? Scand J Immunol (2009) 70(4):326–36. doi:10.1111/j.1365-3083.2009.02308.x
92. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity (2009) 31(2):331–41. doi:10.1016/j.immuni.2009.08.001
93. Johnson AJ, Suidan GL, Chen Y, Pirko I. Rapid up regulation of CNS VEGF is initiated by CD8 T cells in a murine model of BBB disruption. FASEB J (2008) 22(1 Suppl):1072.4.
94. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology (1970) 18(5):723–37.
95. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64.
96. Kitaichi N, Namba K, Taylor AW. Inducible immune regulation following autoimmune disease in the immune-privileged eye. J Leukoc Biol (2005) 77(4):496–502. doi:10.1189/jlb.0204114
97. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi:10.1038/ni904
98. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev (2006) 212(1):8–27. doi:10.1111/j.0105-2896.2006.00427.x
99. Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med (2009) 15(2):192–9. doi:10.1038/nm.1927
100. Stubbe T, Ebner F, Richter D, Engel OR, Klehmet J, Royl G, et al. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab (2013) 33(1):37–47. doi:10.1038/jcbfm.2012.128
101. Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Göbel K, Schuhmann MK, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood (2013) 121(4):679–91. doi:10.1182/blood-2012-04-426734
102. Xu X, Li M, Jiang Y. The paradox role of regulatory T cells in ischemic stroke. ScientificWorldJournal (2013) 2013:174373. doi:10.1155/2013/174373
103. McPherson SW, Heuss ND, Lehman U, Gregerson DS. Generation of regulatory T cells to antigen expressed in the retina. Curr Immunol Rev (2011) 7(3):344. doi:10.2174/157339511796196584
104. Ke Y, Jiang G, Sun D, Kaplan HJ, Shao H. Ocular regulatory T cells distinguish monophasic from recurrent autoimmune uveitis. Invest Ophthalmol Vis Sci (2008) 49(9):3999–4007. doi:10.1167/iovs.07-1468
105. Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J (2009) 276(1):13–26. doi:10.1111/j.1742-4658.2008.06766.x
106. Gesslein B, Håkansson G, Gustafsson L, Ekström P, Malmsjö M. Tumor necrosis factor and its receptors in the neuroretina and retinal vasculature after ischemia-reperfusion injury in the pig retina. Mol Vis (2010) 16:2317–27.
107. Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci (2002) 22(7):1–7.
108. Yoshida S, Yoshida A, Ishibashi T. Induction of IL-8, MCP-1, and bFGF by TNF-α in retinal glial cells: implications for retinal neovascularization during post-ischemic inflammation. Graefes Arch Clin Exp Ophthalmol (2004) 242(5):409–13. doi:10.1007/s00417-004-0874-2
109. Yoshida S, Ono M, Shono T, Izumi H, Ishibashi T, Suzuki H, et al. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol Cell Biol (1997) 17(7):4015–23. doi:10.1128/MCB.17.7.4015
110. Li H-L, Kostulas N, Huang Y-M, Xiao B-G, van der Meide P, Kostulas V, et al. IL-17 and IFN-γ mRNA expression is increased in the brain and systemically after permanent middle cerebral artery occlusion in the rat. J Neuroimmunol (2001) 116(1):5–14. doi:10.1016/S0165-5728(01)00264-8
111. Li GZ, Zhong D, Yang LM, Sn B, Zhong ZH, Yin YH, et al. Expression of interleukin-17 in ischemic brain tissue. Scand J Immunol (2005) 62(5):481–6. doi:10.1111/j.1365-3083.2005.01683.x
112. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201(2):233–40. doi:10.1084/jem.20041257
113. Sanchez RN, Chan CK, Garg S, Kwong JM, Wong MJ, Sadun AA, et al. Interleukin-6 in retinal ischemia reperfusion injury in rats. Invest Ophthalmol Vis Sci (2003) 44(9):4006–11. doi:10.1167/iovs.03-0040
114. Hangai M, Yoshimura N, Yoshida M, Yabuuchi K, Honda Y. Interleukin-1 gene expression in transient retinal ischemia in the rat. Invest Ophthalmol Vis Sci (1995) 36(3):571–8.
115. Berger S, Savitz SI, Nijhawan S, Singh M, David J, Rosenbaum PS, et al. Deleterious role of TNF-α in retinal ischemia-reperfusion injury. Invest Ophthalmol Vis Sci (2008) 49(8):3605–10. doi:10.1167/iovs.07-0817
116. Lupo G, Motta C, Giurdanella G, Anfuso CD, Alberghina M, Drago F, et al. Role of phospholipases A2 in diabetic retinopathy: in vitro and in vivo studies. Biochem Pharmacol (2013) 86(11):1603–13. doi:10.1016/j.bcp.2013.09.008
117. Bucolo C, Maltese A, Maugeri F, Ward KW, Baiula M, Sparta A, et al. New coumarin-based anti-inflammatory drug: putative antagonist of the integrins alphaLbeta2 and alphaMbeta2. J Pharm Pharmacol (2008) 60(11):1473–9. doi:10.1211/jpp.60.11.0008
118. Fisichella V, Giurdanella G, Platania CB, Romano GL, Leggio GM, Salomone S, et al. TGF-beta1 prevents rat retinal insult induced by amyloid-beta (1-42) oligomers. Eur J Pharmacol (2016) 787:72–7. doi:10.1016/j.ejphar.2016.02.002
119. Ganguly K, Giddaluru J, August A, Khan N. Post-transcriptional regulation of immunological responses through riboclustering. Front Immunol (2016) 7:161. doi:10.3389/fimmu.2016.00161
120. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta (2013) 1833(12):3460–70. doi:10.1016/j.bbamcr.2013.06.028
121. Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem (2005) 74:739–89. doi:10.1146/annurev.biochem.73.011303.074134
122. Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res (2005) 569(1):29–63. doi:10.1016/j.mrfmmm.2004.06.056
123. Zhang SX, Sanders E, Fliesler SJ, Wang JJ. Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp Eye Res (2014) 125:30–40. doi:10.1016/j.exer.2014.04.015
124. Doh SH, Kim JH, Lee KM, Park HY, Park CK. Retinal ganglion cell death induced by endoplasmic reticulum stress in a chronic glaucoma model. Brain Res (2010) 1308:158–66. doi:10.1016/j.brainres.2009.10.025
125. Ravindran R, Khan N, Nakaya HI, Li S, Loebbermann J, Maddur MS, et al. Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science (2014) 343(6168):313–7. doi:10.1126/science.1246829
126. Matsui Y, Kyoi S, Takagi H, Hsu C-P, Hariharan N, Ago T, et al. Molecular mechanisms and physiological significance of autophagy during myocardial ischemia and reperfusion. Autophagy (2008) 4(4):409–15. doi:10.4161/auto.5638
127. Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion roles of AMP-activated protein kinase and beclin 1 in mediating autophagy. Circ Res (2007) 100(6):914–22. doi:10.1161/01.RES.0000261924.76669.36
128. Piras A, Gianetto D, Conte D, Bosone A, Vercelli A. Activation of autophagy in a rat model of retinal ischemia following high intraocular pressure. PLoS One (2011) 6(7):e22514. doi:10.1371/journal.pone.0022514
Keywords: retina, ischemia–reperfusion, inflammation, immune response, stress response
Citation: Minhas G, Sharma J and Khan N (2016) Cellular Stress Response and Immune Signaling in Retinal Ischemia–Reperfusion Injury. Front. Immunol. 7:444. doi: 10.3389/fimmu.2016.00444
Received: 19 July 2016; Accepted: 07 October 2016;
Published: 24 October 2016
Edited by:
Kai Fang, University of California Los Angeles, USAReviewed by:
M. Heather West Greenlee, Iowa State University, USAClaudio Bucolo, University of Catania, Italy
Copyright: © 2016 Minhas, Sharma and Khan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nooruddin Khan, noor@uohyd.ac.in