
Enhanced Cellular Uptake and Pharmacokinetic Characteristics of Doxorubicin-Valine Amide Prodrug

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
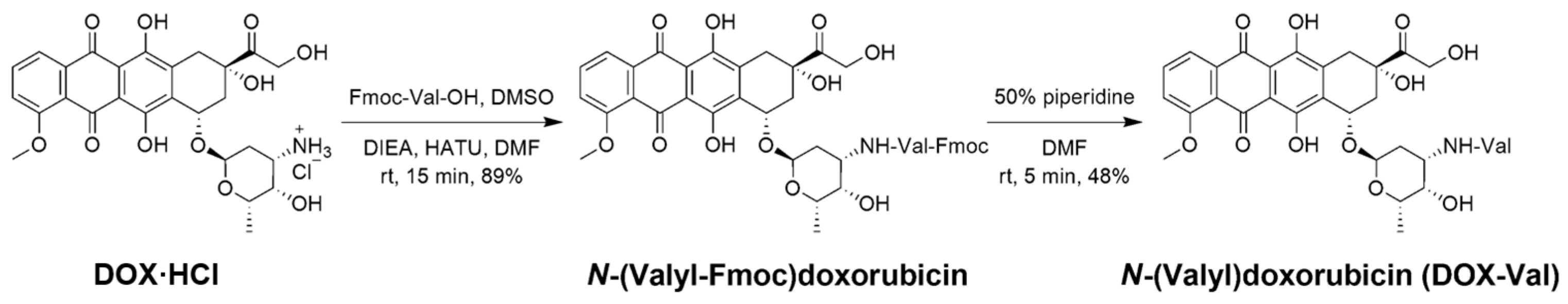
2.1. Synthesis and Characterization of DOX-Val
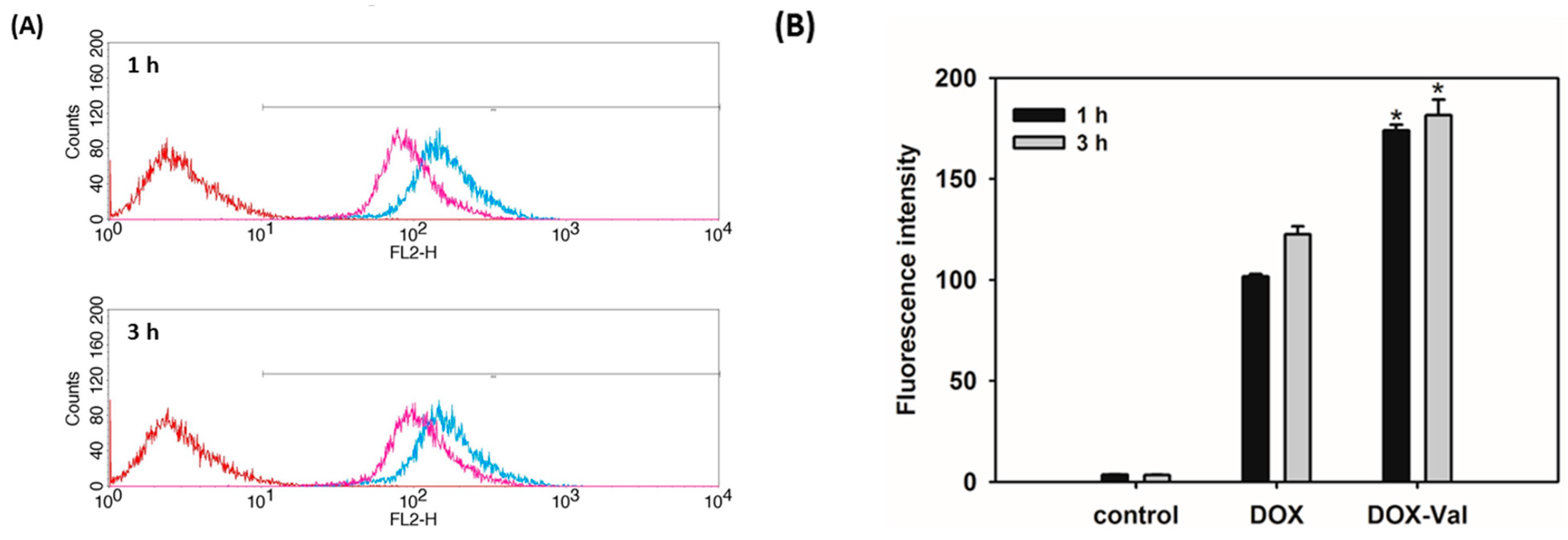
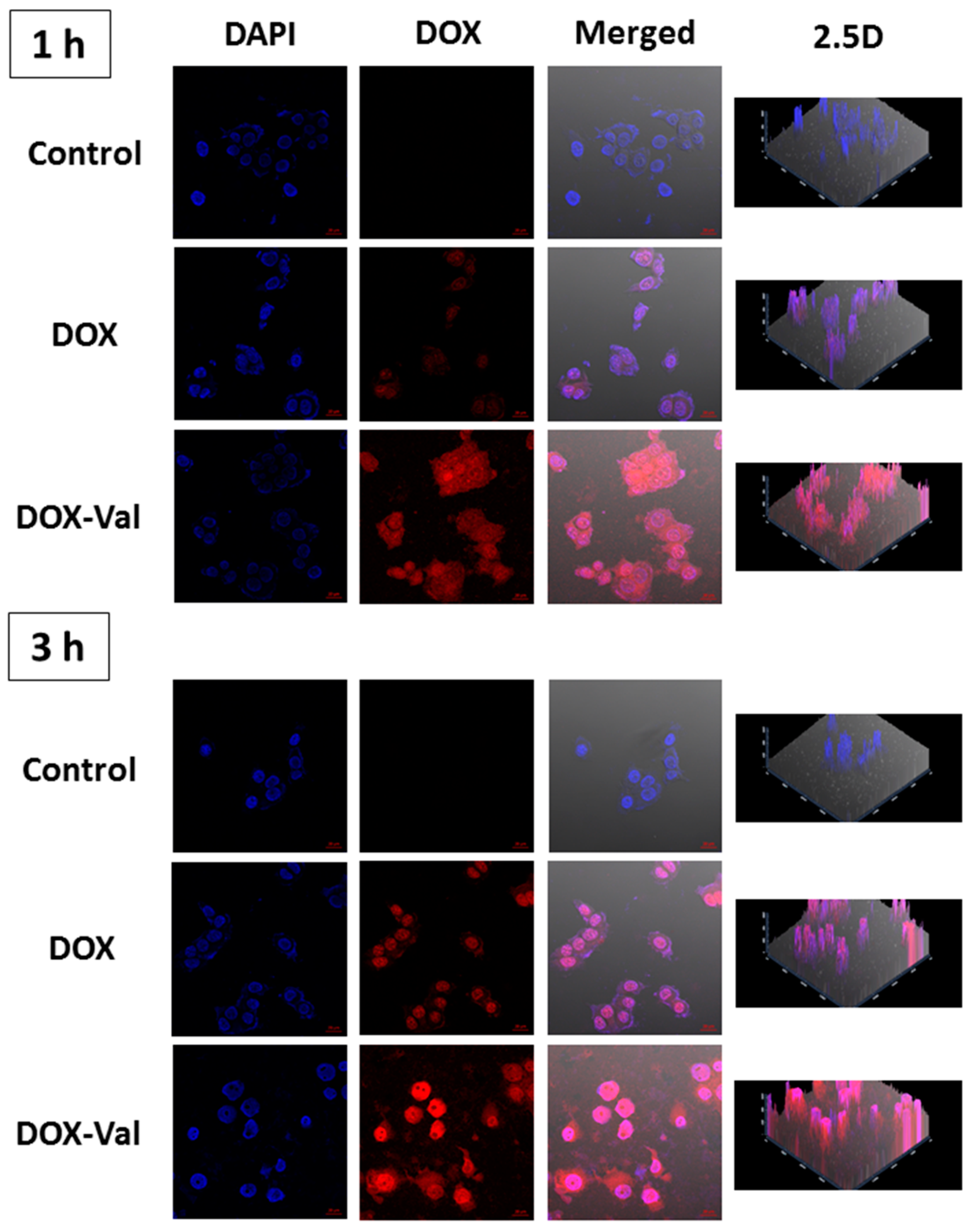
2.2. Cellular Uptake of DOX-Val
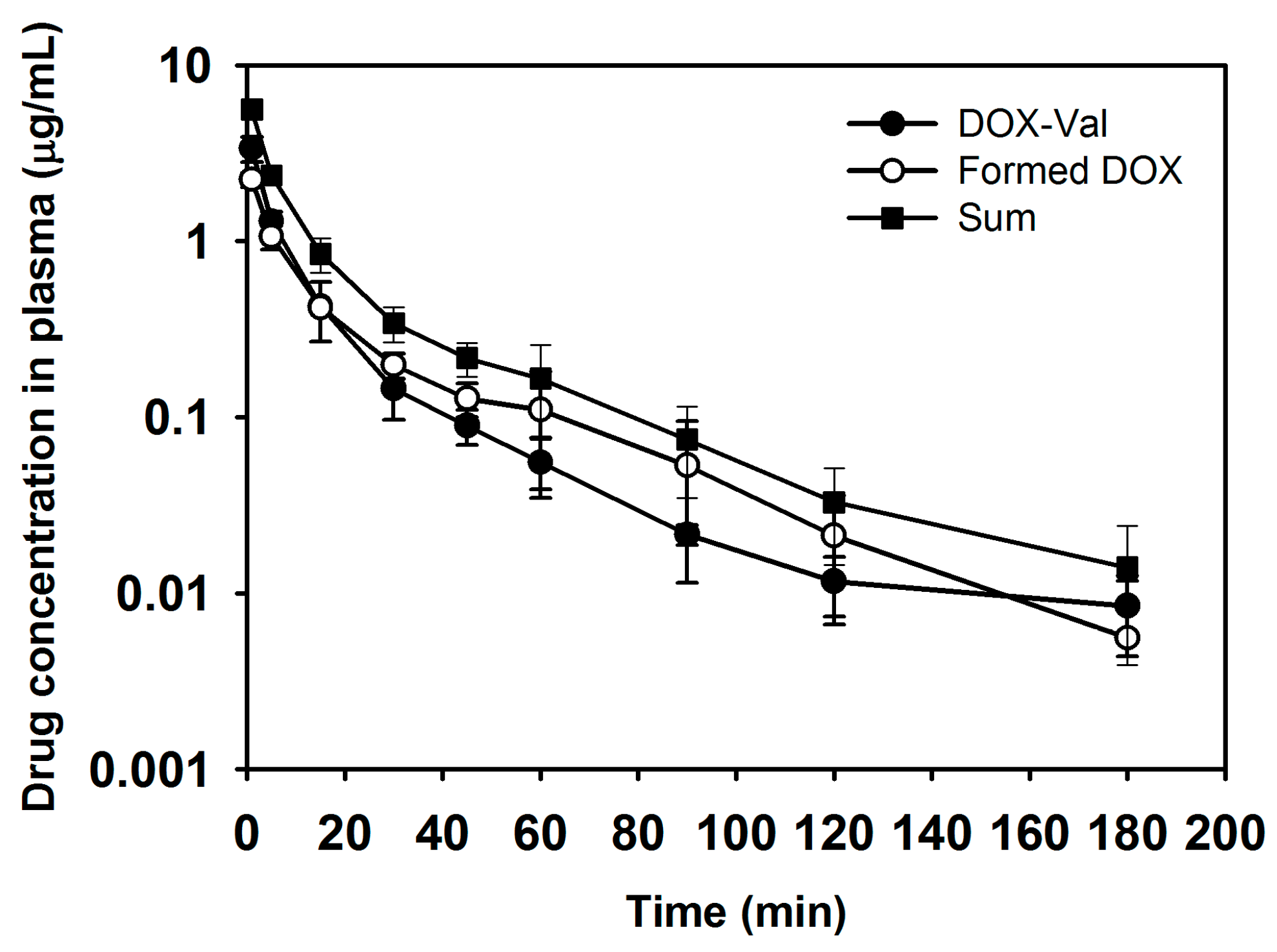
2.3. Pharmacokinetics of DOX-Val
3. Materials and Methods
3.1. Materials
3.2. Synthesis and Characterization of DOX-Val
3.2.1. N-(Valyl-Fmoc)doxorubicin
3.2.2. DOX-Val
3.3. Cellular Uptake Efficiency Study in MCF-7 Cells
3.3.1. Flow Cytometry
3.3.2. CLSM
3.4. Pharmacokinetic Study
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, J.Y.; Chung, S.J.; Cho, H.J.; Kim, D.D. Iodinated hyaluronic acid oligomer-based nanoassemblies for tumor-targeted drug delivery and cancer imaging. Biomaterials 2016, 85, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Pridgen, E.M.; Langer, R.; Farokhzad, O.C. Biodegradable, polymeric nanoparticle delivery systems for cancer therapy. Nanomedicine (Lond.) 2007, 2, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Heller, D.A.; Winslow, M.M.; Dahlman, J.E.; Pratt, G.W.; Langer, R.; Jacks, T.; Anderson, D.G. Treating metastatic cancer with nanotechnology. Nat. Rev. Cancer 2011, 12, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Mandlekar, S.; Marathe, P. Prodrug design to improve pharmacokinetic and drug delivery properties: Challenges to the discovery scientists. Curr. Med. Chem. 2010, 17, 3874–3908. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Dragojevic, S.; Ryu, J.S.; Raucher, D. Polymer-BASED PRODRUGS: Improving tumor targeting and the solubility of small molecule drugs in cancer therapy. Molecules 2015, 20, 21750–21769. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Wang, Y.; Ma, Y.; Zhang, D.; Fallon, J.K.; Yang, X.; Liu, D.; He, Z.; Liu, F. Programmed hydrolysis in designing paclitaxel prodrug for nanocarrier assembly. Sci. Rep. 2015, 5, 12023. [Google Scholar] [CrossRef] [PubMed]
- Gou, Y.; Yang, F.; Liang, H. Designing prodrugs based on special residues of human serum albumin. Curr. Top. Med. Chem. 2016, 16, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lyv, Z.; Li, Y.; Liu, H.; Wang, J.; Zhan, W.; Chen, H.; Chen, H.; Li, X. A theranostic prodrug delivery system based on Pt(IV) conjugated nano-graphene oxide with synergistic effect to enhance the therapeutic efficacy of Pt drug. Biomaterials 2015, 51, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Li, B.B.; Lu, X.; Jia, F.; Santori, C.; Menon, P.; Li, H.; Zhang, B.; Zhao, J.J.; Zhang, K. Light-triggered, self-immolative nucleic Acid-drug nanostructures. J. Am. Chem. Soc. 2015, 137, 6112–6115. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F.; Schmuck, K. Prodrugs for targeted tumor therapies: Recent developments in ADEPT, GDEPT and PMT. Curr. Pharm. Des. 2011, 17, 3527–3547. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F.; Müller, I.A.; Ryppa, C.; Warnecke, A. Prodrug strategies in anticancer chemotherapy. Chem. Med. Chem. 2008, 3, 20–53. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; McLeod, H.L. Strategies for enzyme/prodrug cancer therapy. Clin. Cancer Res. 2001, 7, 3314–3324. [Google Scholar] [PubMed]
- Mahato, R.; Tai, W.; Cheng, K. Prodrugs for improving tumor targetability and efficiency. Adv. Drug Deliv. Rev. 2011, 63, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Bildstein, L.; Dubernet, C.; Couvreur, P. Prodrug-based intracellular delivery of anticancer agents. Adv. Drug Deliv. Rev. 2011, 63, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Sun, J.; Sun, B.; He, Z. Prodrug-based nanoparticulate drug delivery strategies for cancer therapy. Trends Pharmacol. Sci. 2014, 35, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.E.; Jin, H.E.; Hong, S.S. Targeting l-type amino acid transporter 1 for anticancer therapy: Clinical impact from diagnostics to therapeutics. Expert Opin. Ther. Targets 2015, 19, 1319–1337. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.Y.; Shim, W.S.; Chang, J.E.; Chong, S.; Kim, D.D.; Chung, S.J.; Shim, C.K. Enhanced intracellular accumulation of a non-nucleoside anti-cancer agent via increased uptake of its valine ester prodrug through amino acid transporters. Xenobiotica 2012, 42, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Maeng, H.J.; Kim, E.S.; Chough, C.; Joung, M.; Lim, J.W.; Shim, C.K.; Shim, W.S. Addition of amino acid moieties to lapatinib increases the anticancer effect via amino acid transporters. Biopharm. Drug Dispos. 2014, 35, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Diez-Torrubia, A.; García-Aparicio, C.; Cabrera, S.; de Meester, I.; Balzarini, J.; Camarasa, M.J.; Velázquez, S. Application of the dipeptidyl Peptidase IV (DPPIV/CD26) based prodrug approach to different amine-containing drugs. J. Med. Chem. 2010, 53, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Morton, C.L.; Danks, M.K.; Potter, P.M. Proficient metabolism of irinotecan by a human intestinal carboxylesterase. Cancer Res. 2000, 60, 4725–4728. [Google Scholar] [PubMed]
- Mathijssen, R.H.; van Alphen, R.J.; Verweij, J.; Loos, W.J.; Nooter, K.; Stoter, G.; Sparreboom, A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin. Cancer Res. 2001, 7, 2182–2194. [Google Scholar] [PubMed]
- Wu, W.; Dong, Y.; Gao, J.; Gong, M.; Zhang, X.; Kong, W.; Li, Y.; Zeng, Y.; Si, D.; Wei, Z.; et al. Aspartate-modified doxorubicin on its N-terminal increases drug accumulation in LAT1-overexpressing tumors. Cancer Sci. 2015, 106, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, O.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Segawa, H.; Nii, T.; Cha, S.H.; Matsuo, H.; Fukushima, J.; Fukasawa, Y.; et al. Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta 2001, 1514, 291–302. [Google Scholar] [CrossRef]
- Cho, H.J.; Yoon, I.S.; Yoon, H.Y.; Koo, H.; Jin, Y.J.; Ko, S.H.; Shim, J.S.; Kim, K.; Kwon, I.C.; Kim, D.D. Polyethylene glycol-conjugated hyaluronic acid-ceramide self-assembled nanoparticles for targeted delivery of doxorubicin. Biomaterials 2012, 33, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: not available.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacokinetic Parameter | DOX-Val | ||
|---|---|---|---|
| DOX-Val (Prodrug) | Formed DOX (Metabolite) | Sum | |
| AUC (μg∙min/mL) | 29.80 ± 4.11 | 28.58 ± 5.38 | 61.68 ± 8.99 |
| Terminal t1/2 (min) | 49.80 ± 7.83 | 25.59 ± 4.41 | 33.66 ± 5.43 |
| CL (mL/min/kg) | 136.19 ± 19.01 | 143.39 ± 24.25 | 65.83 ± 9.10 |
| Vss (mL/kg) | 3010.60 ± 619.85 | 3617.77 ± 625.60 | 1411.31 ± 245.55 |
| MRT (min) | 22.36 ± 5.06 | 26.35 ± 8.96 | 22.07 ± 6.59 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.; Park, J.-H.; Park, S.; Lee, S.Y.; Cho, K.H.; Kim, D.-D.; Shim, W.-S.; Yoon, I.-S.; Cho, H.-J.; Maeng, H.-J. Enhanced Cellular Uptake and Pharmacokinetic Characteristics of Doxorubicin-Valine Amide Prodrug. Molecules 2016, 21, 1272. https://doi.org/10.3390/molecules21101272
Park Y, Park J-H, Park S, Lee SY, Cho KH, Kim D-D, Shim W-S, Yoon I-S, Cho H-J, Maeng H-J. Enhanced Cellular Uptake and Pharmacokinetic Characteristics of Doxorubicin-Valine Amide Prodrug. Molecules. 2016; 21(10):1272. https://doi.org/10.3390/molecules21101272
Chicago/Turabian StylePark, Yohan, Ju-Hwan Park, Suryeon Park, Song Yi Lee, Kwan Hyung Cho, Dae-Duk Kim, Won-Sik Shim, In-Soo Yoon, Hyun-Jong Cho, and Han-Joo Maeng. 2016. "Enhanced Cellular Uptake and Pharmacokinetic Characteristics of Doxorubicin-Valine Amide Prodrug" Molecules 21, no. 10: 1272. https://doi.org/10.3390/molecules21101272