Article Text

Abstract
Objective The recent availability of novel antiviral drugs has raised new hope for a more effective treatment of hepatitis C virus (HCV) infection and its severe sequelae. However, in the case of non-responding or relapsing patients, alternative strategies are needed. To this end we have used chimeric antigen receptors (CARs), a very promising approach recently used in several clinical trials to redirect primary human T cells against different tumours. In particular, we designed the first CARs against HCV targeting the HCV/E2 glycoprotein (HCV/E2).
Design Anti-HCV/E2 CARs were composed of single-chain variable fragments (scFvs) obtained from a broadly cross-reactive and cross-neutralising human monoclonal antibody (mAb), e137, fused to the intracellular signalling motif of the costimulatory CD28 molecule and the CD3ζ domain. Activity of CAR-grafted T cells was evaluated in vitro against HCV/E2-transfected cells as well as hepatocytes infected with cell culture-derived HCV (HCVcc).
Results In this proof-of-concept study, retrovirus-transduced human T cells expressing anti-HCV/E2 CARs were endowed with specific antigen recognition accompanied by degranulation and secretion of proinflammatory and antiviral cytokines, such as interferon γ, interleukin 2 and tumour necrosis factor α. Moreover, CAR-grafted T cells were capable of lysing target cells of both hepatic and non-hepatic origin expressing on their surface the HCV/E2 glycoproteins of the most clinically relevant genotypes, including 1a, 1b, 2a, 3a, 4 and 5. Finally, and more importantly, they were capable of lysing HCVcc-infected hepatocytes.
Conclusions Clearance of HCV-infected cells is a major therapeutic goal in chronic HCV infection, and adoptive transfer of anti-HCV/E2 CARs-grafted T cells represents a promising new therapeutic tool.
- HEPATITIS C
- IMMUNOLOGY IN HEPATOLOGY
- IMMUNOTHERAPY
- INFECTIOUS DISEASE
- IMMUNE RESPONSE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Hepatitis C virus (HCV) is a leading cause of chronic liver disease worldwide.
Recently approved direct-acting anti-HCV drugs show promising results when compared to the previous standard of care consisting of the administration of interferon α (IFN-α) and ribavirin.
The occurrence of viral escape mechanisms and resistance, as well as side effects of the novel drugs are still potential therapeutic concerns.
Alternative therapeutic strategies that can be employed in selected categories of chronically infected patients (ie, non-responders; end-stage patients) may therefore still be needed.
What are the new findings?
Chimeric antigen receptors (CARs) represent a very promising approach that, besides being useful in oncology, can also be adopted in infectious diseases.
Two anti-HCV CARs were designed based on a previously described broadly cross-reactive and cross-neutralising human monoclonal antibody (mAb), named e137, directed against a conserved epitope of the HCV E2 glycoprotein (HCV/E2).
Anti-HCV CAR-grafted T cells are able to recognise and lyse target cells expressing HCV/E2 of different genotypes and subtypes. In particular, anti-HCV CAR-grafted T cells showed cytotoxic activity against HCV/E2-transfected as well as HCV cell culture (HCVcc)-infected target cells. Moreover, antigen recognition was accompanied by T cell degranulation and the secretion of proinflammatory and antiviral cytokines.
How might it impact on clinical practice in the foreseeable future?
In this proof-of-concept study we have shown that T cells grafted with CARs directed against HCV/E2 could represent an alternative strategy that could be adopted in cases where patients are unresponsive to available therapies.
Introduction
Hepatitis C virus (HCV) is one of the major causes of chronic liver disease, with about 180 million people infected worldwide, and therefore exposed to the risk of chronic liver complications, such as cirrhosis, liver failure and hepatocellular carcinoma.1–4 In recent years, antiviral therapies have rapidly evolved with the development of licensed anti-HCV molecules, including viral protease and polymerase inhibitors, in addition to the previous standard of care consisting of the administration of interferon α (IFN-α) and ribavirin.5 However, as observed in other chronic infections, such as HIV and HBV, several factors can hamper the effectiveness of these novel molecules, such as the occurrence of resistance, the incomplete compliance to the treatment regime and as yet unknown side effects.6 ,7
In fact, as with other viruses causing a chronic infection, one of the main features of HCV is its capacity to rapidly mutate under selective pressure, such as the host immune response or ongoing therapy.8 Incidentally, chronic HCV infection is the major determinant for liver transplantation, being the only available treatment in end-stage chronically infected patients not responding to current therapies.9 Unfortunately, post-transplant liver re-infection is almost universal. The degeneration of the transplanted liver is usually more dramatic and rapid, and its treatment is even more problematic than in the pretransplant setting.9 Despite recent therapeutic novelties, all of the above make the need for other prophylactic and therapeutic approaches even more compelling.
In most cases, HCV is capable of subverting the host immune response by using a plethora of different escape mechanisms. HCV E2 glycoprotein (HCV/E2) is the major target of the host immune response and, as a consequence, it is also the most variable viral protein, giving rise to highly diversified variants (quasispecies).10 In fact, HCV/E2 is, together with E1, the major surface envelope protein, and includes in its structure the receptor binding domains and the putative fusion loop, essential regions for the viral entry process.10 ,11 HCV/E1 and E2 are both believed to be retained in the endoplasmic reticulum or cis-Golgi compartment but, importantly for the approach proposed in this paper, they have also been detected at the level of the cell surface of transfected cells12 and, as supported by our data, of infected hepatocytes. Moreover, the functional evidence that some anti-HCV/E2 monoclonal antibodies (mAbs) are able to reduce the HCV cell-to-cell route of transmission supports the localisation and targeting of this glycoprotein also at the level of the cell surface.13 ,14
There is growing evidence that a strong and broad HCV-specific CD4+ and CD8+ T cell response, as well as a broadly cross-neutralising humoral immunity, are required for viral clearance.15 In this respect, HCV has evolved a number of mechanisms to elude the host immune response. It is well noted that HCV infection leads to an impaired innate immune response and, consequently, to the shaping of a less effective adaptive response.16 Moreover, major histocompatibility complex (MHC) downregulation and mutations within CD4+ and CD8+ T cell HCV epitopes are considered to be among the most important mechanisms leading to the failure of T cell-mediated immune response, as observed in chronically infected patients.15
As far as the humoral immune response is concerned, it is well documented that rarely elicited cross genotype-neutralising antibodies are associated to the resolution of acute infection.17 ,18 Several anti-HCV/E2 broadly cross-neutralising mAbs have been described and are ready to be tested also in clinical trials, such as e137 used in this study.10 ,19–23 It should be noted, however, that in the case of the humoral immune response, several viral escape mechanisms have also been described. In particular, viral escape variants capable of evading the humoral response have been widely studied, mostly at the level of the HCV/E2 glycoprotein.24 Among other escape mechanisms, the masking of crucial HCV/E2 regions involved in the binding and entry into target cells, as well as the induction of non-neutralising/interfering antibodies and hidden cell-to-cell transmission, have all been well described.25–30 In particular, this last route of evasion has also been described as a resistance mechanism for the recently approved direct-acting antivirals.31
Thus, the correct shaping of both the humoral and cell-mediated immune response could constitute an effective strategy for the clearance of the infection. To this end, the adoptive transfer of T cells directed against viral antigens and the subsequent elimination of infected cells could represent a valid support strategy for currently available therapies. Recently, Zhang et al32 isolated from a chronically infected patient an HLA-A2-restricted, cytotoxic T cell clone and expressed its anti-HCV/NS3 TCR in human T cells. Functional analysis of the resulting TCR-transduced cells evidenced that the CD4+ and CD8+ T cells both recognised HCV/NS3-espressing hepatocytes with cytokine production and cytotoxic activity.32 A possible MHC-independent alternative could be the use of chimeric antigen receptors (CARs) to arm T cells against highly conserved HCV antigens expressed on the surface of infected cells. Using this recently developed technology, every desired cell-surface exposed antigen could ideally be targeted, exploiting the antibody specificity given by single-chain variable antibody fragments (scFvs) cloned in frame with the intracellular signal machinery of the T cell receptor (TCR) and of other co-stimulatory regions.33 Thus, CAR T cell engineering could possibly avoid escape mechanisms involving MHC-dependent antigen presentation or downregulation.33 In a similar way, other HCV escape mechanisms from humoral immune response, such as cell-to-cell transmission, may be circumvented.
In this paper, we describe for the first time the engineering of healthy donor T cells with CARs targeting a highly conserved epitope of HCV/E2 glycoprotein. In particular, these CARs feature a broadly cross-reactive profile conferred by a previously described broadly cross-reacting and cross-neutralising human mAb, named e137. This mAb targets with high affinity an extremely conserved region of the HCV/E2 glycoprotein, of which the aminoacid residues are involved in the binding to the CD81 receptor and are crucial for HCV infectivity, and it is able to broadly recognise and neutralise different HCV genotypes and subtypes.19 ,21 ,34 Moreover, its activity is not influenced by the presence of possible non-neutralising/interfering antibodies.25 It is important to highlight that non-neutralising mAbs could also be engineered as CARs on the basis of their binding characteristics. However, non-neutralising mAbs are usually directed against functionally less important, and therefore more variable epitopes, thus potentially decreasing the broadness of action of the resulting CARs against possible escape variants.25 ,35 Although not impossible, this event is certainly less probable when using broadly neutralising mAbs directed against functionally pivotal HCV/E2 epitopes, such as e137, used in this study.19
In this proof-of-concept study, we demonstrate that CAR-mediated targeting of HCV/E2 glycoprotein is a promising approach to confer a sterilising clearance of HCV-infected cells, opening possible future perspectives in the treatment of patients not responding to current antiviral treatments or in those undergoing liver transplantation.
Materials and methods
CAR construction and plasmid preparation
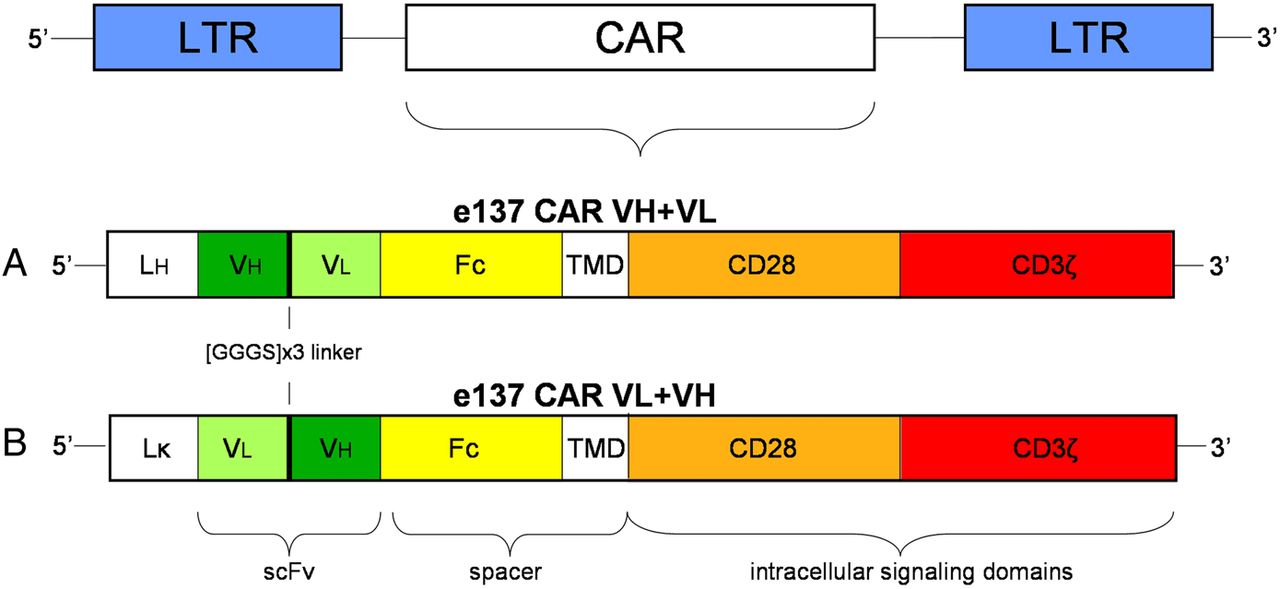
Anti-HCV/E2 mAb e137 was selected from a phage-displayed antibody library of an HCV genotype 1b-chronically infected patient, as previously described.19 ,35–38 DNA encoding e137 scFv composed of the light chain variable (VL) domain followed by a [GGGS]x3 linker and the heavy chain variable (VH) domain sequence (named VL+VH), or of the VH domain followed by the linker and the VL domain (VH+VL), were cloned at the 3′-end of the Vκ1-9 or the VH1-69 leader sequences, respectively. Constructs were inserted at the 5′-end of a human IgG-Fc spacer region followed by the sequences of the CD28 intracellular signalling motif and of the CD3ζ domain in a second generation MP71 retroviral vector for the expression of the related CARs (e137-CARs).39 Figure 1 shows the schematic representation of the regions composing the engineered e137-CARs.

Schematic representation of the chimeric antigen receptor (CAR) constructs inserted into the retroviral vector plasmid MP71. (A) e137 VH+VL is composed of a N-terminal heavy-chain leader sequence (LH), with the VH domain followed by the VL domain, whereas (B) e137 VL+VH features a N-terminal light κ-chain leader sequence (Lκ), with the VL domain followed by the VH domain. Both e137-derived scFvs are inserted at the 5′-end of a human IgG1 Fc spacer domain (Fc), transmembrane and intracellular regions of the CD28 signalling domain (CD28) and the CD3ζ signalling domain (CD3ζ). LTR, long terminal repeat.
The previously described S-CAR vector, expressing a CAR directed against the hepatitis B virus S antigen (HBsAg) was used as positive control for retroviral transduction, and as negative control in cytotoxicity assays of transduced T cells cocultured with the different target cells.40 All vectors were amplified using the XL1-Blue strain (Stratagene, La Jolla, California, USA) and purified with a Plasmid MIDI Kit (Qiagen, Venlo, The Netherlands). All the CAR constructs were grafted into T cells derived from three different healthy donors, as described in the online supplementary methods and figure S1.
T-cell activation and cytotoxicity assays on transiently transfected target cells
Untransfected and transiently transfected HEK-293T cells (generated as described in the online supplementary methods) with plasmids containing the HCV/E1–E2 sequences of isolates belonging to different HCV genotypes and subtypes were plated (5×104 cells/well) on a 96-well plate (Costar, New York, USA) until confluency. Analogously, HEK-293T cells transiently transfected with single-point mutated HCV/E1–E2 (T416A, S419A, W420A, W529A, G530A, D535A and V538A),19 ,41 or with the haemagglutinin (HA) sequence of influenza A/Puerto Rico/8/1934 strain were used as controls.
Untransduced, S-CAR-redirected and e137-CARs-redirected total T cells were added in triplicate at a single effector to target (E:T) 1:4 ratio, and co-cultured in IL-2-free medium for 72 h. As complete lysis control, target cells were incubated with 2% (vol/vol) of Triton X-100 (Sigma-Aldrich), while no lysis was achieved by incubating target cells in the absence of effector cells. Supernatants were then collected and target cells washed twice with phosphate-buffered saline (PBS). Specific cytotoxicity of CAR-grafted T cells was determined by an XTT-based colourimetric assay (Cell Proliferation Kit II 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide XTT, Roche, Basel, Switzerland), following the instructions provided by the manufacturer.
T-cell activation and cytotoxicity assays on stably transfected target cells
Untransfected (HEK-293 cells stably transfected with a control protein were not available) and stably transfected HEK-293 cells with HCV/E1–E2 (H77 strain, genotype 1a) or soluble HCV/E2 glycoprotein (sE2, H77 strain), generated as described in the online supplementary methods, were plated (5×104 cells/well) on a 96-well plate (Costar) until confluency. Similarly, stably transfected HepG2 cells expressing HCV/E1–E2 (H77 strain), HCV/sE2 (H77 strain) or transduced with human CD81 (generated as described in the online supplementary methods), were plated (5×104 cells/well) on a collagen type I-coated 96-well plate in medium supplemented with 1% dimethyl sulfoxide (DMSO) (Sigma-Aldrich) to obtain proper differentiation, and cultured until confluency.42 Untransduced (used at a single 1:4 effector to target (E:T) ratio) and four different twofold dilutions of control S-CAR-redirected or e137-CARs-redirected total T cells were then added in triplicate at different E:T ratios (starting from 1:4), and co-cultured in interleukin 2 (IL-2)-free medium for 48 h. The same experiments were also performed using sorted CD4+ or CD8+ CAR-transduced and untransduced T cells, isolated as described in the online supplementary methods. Lysis controls were performed as above.
Supernatants were then collected and target cells washed twice with PBS. Specific cytotoxicity of CARs-grafted T cells against target cells were evaluated analysing target cells as described above.
In all of the above experimental settings, supernatants were removed and analysed with Human IFN-γ, IL-2, TNF-α and Granzyme B ELISA MAX Standard kits (BioLegend Inc, San Diego, California, USA) for IFN-γ, IL-2, TNF-α and granzyme B quantification, respectively, following the instructions provided by the manufacturer.
T-cell activation and cytotoxicity assays on HCVcc-infected target cells
For cytotoxicity assays on JFH-1 strain, HCVcc-infected HuH-7.5 hepatoma cells were used. Briefly, 5×104 HuH-7.5 cells/well were plated in a 96-well plate (Costar), infected at least 48 h before co-culture experiments with high titre HCVcc particles (100 focus forming units (FFU) per well), and incubated until confluency. The number of infected cells was determined using a mouse mAb (9E10 clone, Apath LLC, New York, USA) directed against the HCV/NS5A non-structural protein, and only cultures showing an 80% of HCV/E2-expression were used in the cytotoxic assays. HCV/E2 expression on cell surface was evaluated using e137 mAb on non-permeabilised cells, as reported in the online supplementary methods. Subsequently, untransduced T cells (used at a single 1:4 E:T ratio) and four different twofold dilutions of S-CAR-redirected and e137-CARs-redirected total T cells were added in triplicate at different effectors to target (E:T) ratios starting from 1:4, and co-cultured in IL-2-free medium for 48 h. The same experiments were also performed using sorted CD4+ or CD8+ CAR-transduced and untransduced T cells. Supernatants and target cells were then analysed as described above.
Statistical analysis
All results of cytotoxic assays and IFN-γ, IL-2, TNF-α and granzyme B secretion levels are reported as percentages and absolute mean values, respectively. IFN-γ, IL-2, TNF-α and granzyme B levels secreted by CAR-redirected T cells or controls were determined comparing sample OD values with standard OD values through a non-linear regression analysis. Different raw data groups were compared using the non-parametric Kruskal-Wallis test, considering statistically significant a p value <0.05. All statistical analysis was performed using GraphPad Prism V.5.00 software (San Diego, California, USA).
Results
e137-CARs redirected T cells are activated by coated HCV/E2 glycoprotein and not by free HCV/E2 antigen in the medium
To test e137-CARs activation, transduced T cells were incubated in the presence of coated HCV/E2 glycoprotein, or bovine serum albumin (BSA) as control antigen. A specific activation of e137-CAR VH+VL-transduced CD4+ and CD8+ T cells was evidenced by secretion of IFN-γ, IL-2 and TNF-α, as well as by degranulation-associated CD107a (lysosome-associated membrane protein 1, LAMP-1) cell-surface translocation, but only in the presence of HCV/E2 and not of BSA (figure 2 and online supplementary figures S2 and S3). Similar results were obtained using e137-CAR VL+VH-transduced T cells (data not shown), whereas untransduced or S-CAR-transduced T cells did not show any relevant activation profile (figure 2 and online supplementary figures S2 and S3).

Intracellular staining for interferon γ (IFN-γ), tumour necrosis factor α (TNF-α) and interleukin 2 (IL-2) of engineered T cells redirected with e137-CARs and cultured in presence of coated antigens. The percentages of e137-CAR VH+VL-transduced CD4+ (upper panel) and CD8+ (lower panel) T cells-secreting cytokines are reported. Results are represented as histogram and the mean of percentages plus SE from the mean (error bars) are reported. CAR, chimeric antigen receptor.
Quantification of IFN-γ, IL-2, TNF-α and granzyme B levels confirmed the specificity and the statistical significance of e137-CARs activation by coated HCV/E2 compared to that observed with S-CAR and untransduced T cells (IFN-γ and granzyme B: p<0.01; IL-2 and TNF-α: p<0.05; see online supplementary figure S4).
In order to evaluate the possible activation by the free antigen, e137-CARs-transduced T cells and controls were incubated in medium supplemented with HCV/sE2 or HCVcc viral-particles. For both antigens, e137 CARs-transduced T cells featured significantly lower IFN-γ, IL-2, TNF-α and granzyme B levels than those observed with the coated antigen (IFN-γ: p<0.01; IL-2, TNF-α and granzyme B: p<0.05; see online supplementary figure S4).
e137-CARs grafted T cells are activated by HCV/E1–E2 of different genotypes and subtypes
In order to test their specific cytotoxic activity, CARs-transduced T cells were co-cultured with HEK-293T cells transiently transfected with HCV/E1–E2 constructs derived from eight different HCV genotypes and subtypes, as well as with single-point mutated HCV/E1–E2 variants of H77 strain. In particular, most of these mutants were previously described to abrogate e137 mAb binding and to be crucial for CD81 binding and HCV pseudoparticles infectivity.19 ,41
A cytotoxic effect was observed against almost all subtypes, with an average target clearance of 70% and 71% for genotype 1a (H77 strain), 60% and 70% for genotype 1b (UKN1B12.16 strain), 40% and 50% for genotype 2a (UKN2A1.2), 56% and 62% for genotype 3a (UKN3A1.28 strain), 45% and 55% for genotype 4 (UKN4.21.16 strain), 40% and 41% for genotype 5 (UKN5.15.11 strain), using e137 VH+VL and VL+VH CAR-transduced T cells, respectively (figure 3A). This activity was significantly higher than that observed with untransduced or control S-CAR-redirected T cells, as well as higher than the clearance observed for untrasfected and HA-transfected control target cells (p<0.05 even in the worst case of genotype 5; figure 3A). No significant activity was observed against genotype 2b (UKN2B2.8 strain)-expressing and genotype 6 (UKN6.5.8 strain)-expressing target cells (p>0.05; figure 3A). Moreover, no significant cytotoxic activity was observed against HCV/E1–E2 alanine mutants T416A, W420A, W529A, G530A and D535A previously described to abrogate e137 mAb binding,19 with clearance comparable to that observed on untransfected HEK-293T cells (p>0.05; figure 3B). Conversely, significant cytotoxic activity was observed against S419A and V538A HCV/E1–E2 mutants, previously described not to abrogate e137 mAb binding (p<0.05; figure 3B).19

(A) Cytotoxic activity of engineered T cells redirected with e137-CARs against HEK-293T target cells transiently transfected with HCV/E1–E2 glycoprotein of different genotypes. For each HCV/E1–E2 genotype, the following strains were used: genotype 1a, isolate H77; genotype 1b, isolate UKN1B12.16; genotype 2a, isolate UKN2A1.2; genotype 2b, isolate UKN2B2.8; genotype 3a, isolate UKN3A1.28; genotype 4, isolate UKN4.21.16; genotype 5, isolate UKN5.15.11; genotype 6, isolate UKN6.5.8. Untransfected (mock) and transiently transfected cells with haemagglutinin from A/PR/8/34 influenza strain were used as negative control target cells. Transiently transfected and untransfected HEK-293T cells were co-cultured in triplicate for 72 h with T cells derived from three different healthy donors and grafted with e137-CARs (VH+VL and VL+VH) or control S-CAR, and with untransduced T cells at a 1:4 effector to target (E:T) ratio. Cytotoxic activity is expressed as a percentage of target cells clearance as depicted on y axis. The mean plus SE from the mean (error bars) are reported. (B) Cytotoxic activity of CAR-engineered T cells against HEK-293T cells transiently transfected with HCV/E1–E2 alanine mutants of genotype 1a (H77 strain). On the χ axis the single-point mutated HCV/E1–E2 constructs are reported. Untransfected cells (mock) were used as negative control for target cell clearance. Transiently transfected and untransfected HEK-293T cells were co-cultured in triplicate for 72 h with T cells derived from three healthy donors and grafted with e137-CARs (VH+VL and VL+VH) or control S-CAR, and with untransduced T cells at a single 1:4 E:T ratio. Cytotoxic activity is expressed as a percentage of target cell clearance. The mean plus SE from the mean (error bars) are reported. CAR, chimeric antigen receptor.
To confirm the specific cytotoxic activity, e137-CARs-redirected total and sorted CD4+/CD8+ T cells, as well as total and sorted controls (S-CAR-redirected and untransduced T cells), were co-cultured with HEK-293 cells stably expressing HCV/E1–E2 of genotype 1a (H77 isolate; see online supplementary figure S5A) at different E:T ratios (figure 4A, C, D). Although at the lower limits of statistical significance (p=0.051), e137-CAR VH+VL-transduced and e137-CAR VL+VH-transduced total T cells lysed HCV/E1–E2-expressing target cells, with an average 40% of maximum target clearance at 1:8 E:T ratio (figure 4A). The cytotoxic effect was markedly more evident after T-cell sorting. e137-CAR VH+VL-transduced and e137-CAR VL+VH-transduced CD8+ T cells significantly lysed HCV/E1–E2-expressing target cells, compared to untransduced and S-CAR-transduced T cells, with an average 89% and 88% of maximum target clearance at 1:4 E:T ratio, respectively, and showing a dose-dependent effect at lower E:T ratios (p<0.05; figure 4D).
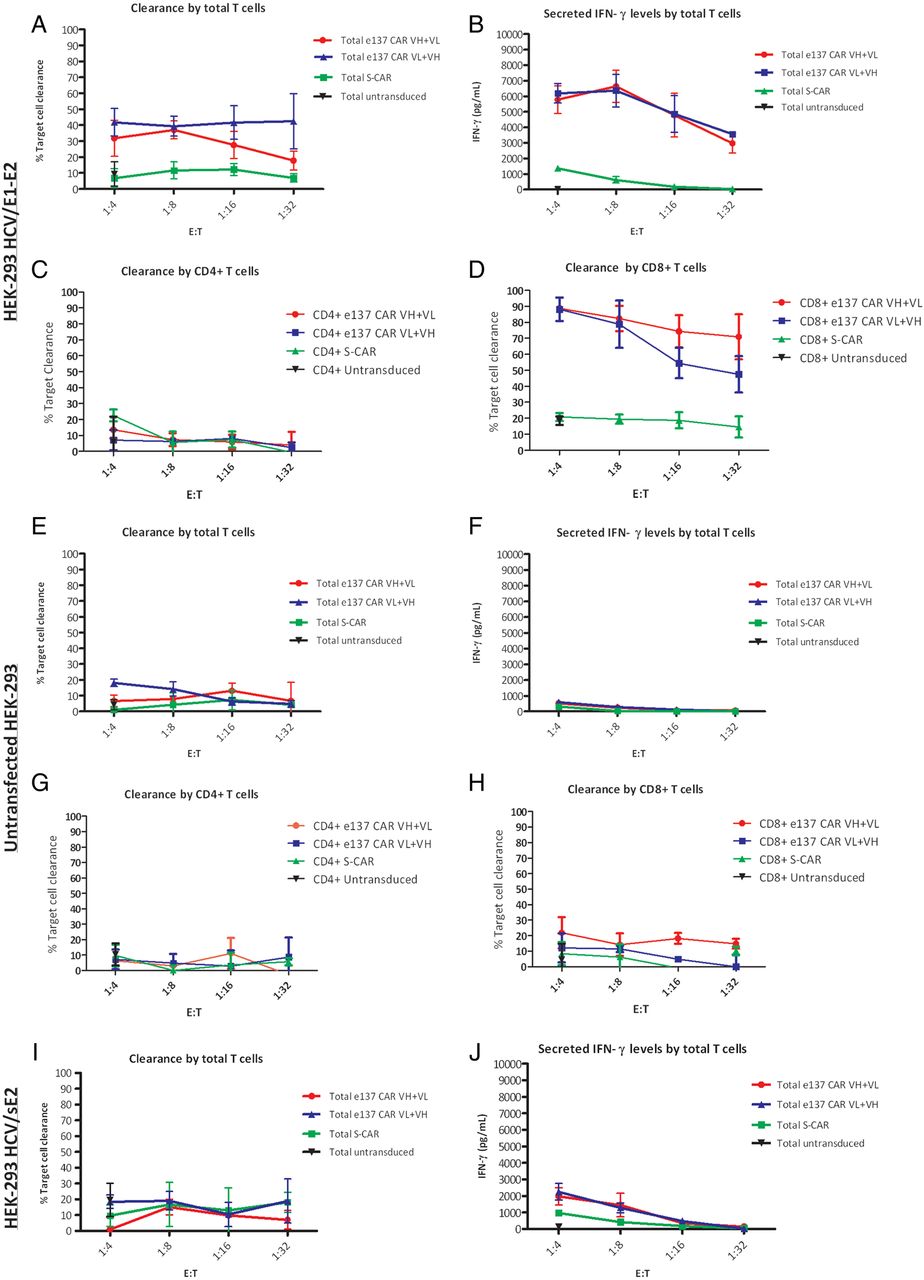
Cytotoxic activity of total and sorted CD4+ and CD8+ engineered T cells redirected with e137-CARs against stably transfected HEK-293 target cells. Panels show the cytotoxic activity (expressed as percentage of target cells clearance) of total and sorted CD4+ and CD8+ T cells derived from three healthy donors and redirected with e137-CARs (VH+VL and VL+VH) or S-CAR against HEK-293 target cells stably transfected with HCV/E1–E2 glycoprotein of genotype 1a (H77 strain; A, C and D), untransfected (E, G and H) or stably transfected with soluble HCV/E2 (HCV/sE2) of genotype 1a (H77 strain) (I). Untransduced total and sorted CD4+ and CD8+ T cells from the same donors were also used as control. Target cells were co-cultured in triplicate for 48 h with CAR-transduced T cells at four different effector to target (E:T) ratios, whereas untransduced T cells were used at a single 1:4 E:T ratio. Panels B, F and J show the corresponding secreted interferon γ (IFN-γ) levels obtained from the different co-cultured total T cells against HEK-293 target cells stably transfected with HCV/E1–E2 glycoprotein, untransfected, or stably transfected with HCV/sE2, respectively. Levels of secreted HCV/sE2 (about 280 ng/mL of medium) were determined as described in the online supplementary methods. The mean plus SE from the mean (error bars) are reported. CAR, chimeric antigen receptor.
Importantly, the activation of total and sorted e137-CARs-transduced T cells was confirmed in a ratio-dependent manner by the significantly higher secreted levels of IFN-γ, IL-2, TNF-α (for total and sorted CD4+/CD8+ T cells) and granzyme B (for total and sorted CD8+ T cells) compared, at each E:T ratio, to controls (IFN-γ and IL-2: p<0.01; TNF-α and granzyme B: p<0.05; figure 4B and online supplementary figure S6). Moreover, confirming what was observed above, the levels of proinflammatory cytokines and granzyme B secreted by e137-CARs-transduced T cells in the presence of HEK-293 cells stably expressing a soluble form of the HCV/E2 (sE2; see online supplementary figure S5A) were significantly lower than those secreted in the presence of stably expressing membrane-bound HCV/E1–E2 HEK-293 cells (p<0.01; figure 4J and online supplementary figure S6). These levels were comparable to those observed with control S-CAR-redirected and untransduced T cells on all target cells (p>0.05; figure 4B, F, J and online supplementary figure S6). Consequently, no significant differences in cytotoxic activity were observed for both total and sorted CD8+ e137-CARs-redirected and control T cells, against HCV/sE2-expressing and untransfected HEK-293 cells (p>0.05; figure 4E, H, I).
e137-CARs grafted T cells are activated by HCV/E1–E2 expressed on the surface of hepatocytes
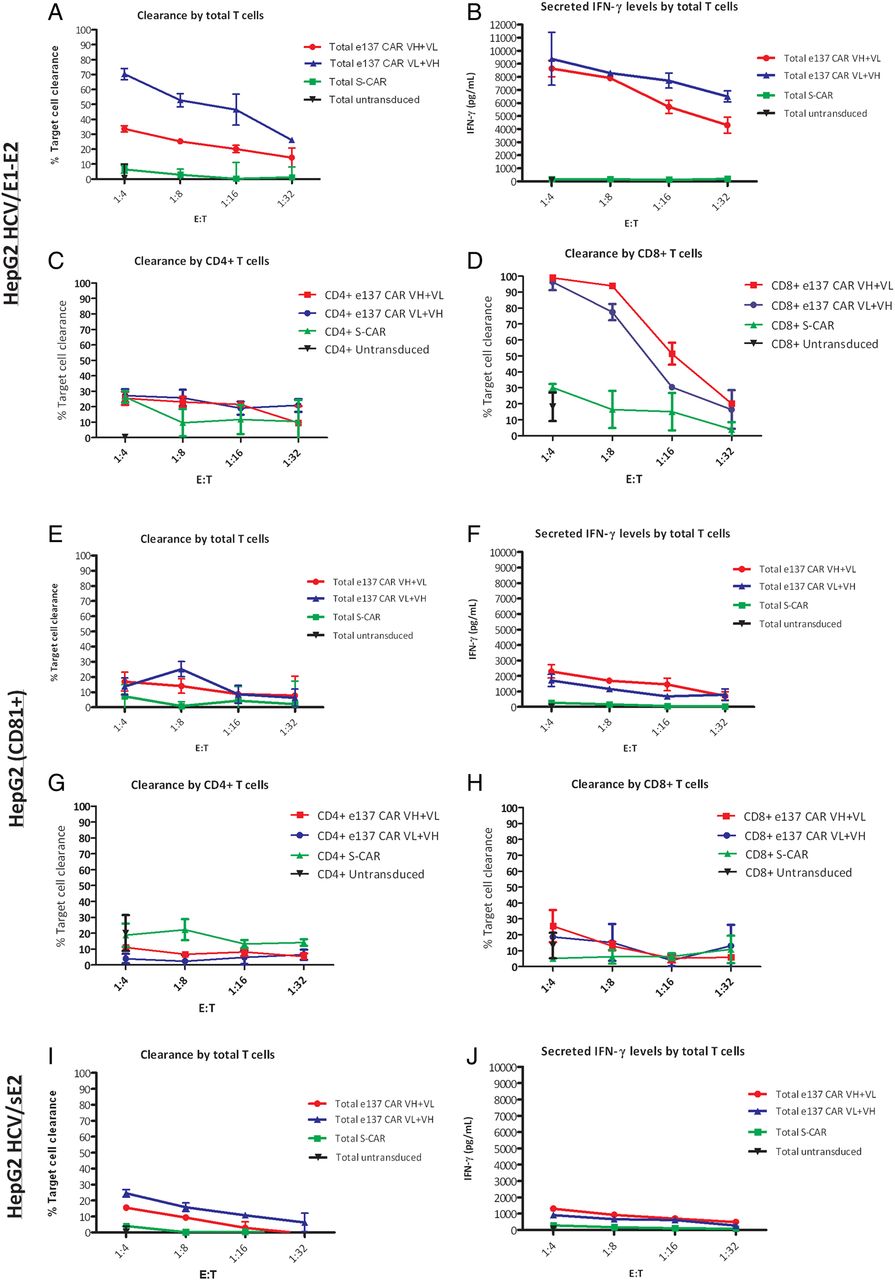
In order to evaluate the cytotoxic activity on hepatocyte-derived cell lines, HepG2 hepatoma cells stably expressing HCV/E1–E2 of genotype 1a (H77 isolate), HCV/sE2 or an unrelated control protein (human CD81) were used as target cells (see online supplementary figure S5B). Cytotoxicity assays confirmed the specific activity of total and sorted CD8+ e137-CAR VH+VL-transduced and e137-CAR VL+VH-transduced T cells also against HCV/E1–E2 stably transfected HepG2 cells (p<0.05). An average of 35% and 70% of maximum target clearance at a 1:4 E:T ratio was observed with e137-CAR VH+VL-transduced and e137-CAR VL+VH-transduced total T cells, respectively (figure 5A). A significantly higher average of 99% and 95% of maximum target clearance at a 1:4 E:T ratio was observed with e137-CAR VH+VL-transduced and e137-CAR VL+VH-transduced CD8+ T cells, respectively (figure 5D and online supplementary figure S7). Importantly, in both settings, a dose-dependent effect at lower E:T ratios was also observed (figure 5A,D). Notably, the evaluation of proinflammatory cytokines and granzyme B levels confirmed the specific activation by HCV/E1–E2-stably transfected HepG2 cells of e137-CARs-redirected compared to control S-CAR-redirected T cells (p<0.01; figure 5B and online supplementary figure S8). Only minimal differences in cytotoxic activity and levels of proinflammatory cytokines and granzyme B were observed for both e137-CARs and controls, against HCV/sE2-transduced and CD81-transduced HepG2 cells (p>0.05; figure 5E, F, H–J and online supplementary figures S7 and S8).

Cytotoxic activity of total and sorted CD4+ and CD8+ engineered T cells redirected with e137-CARs against stably transfected HepG2 target cells. Panels show the cytotoxic activity (expressed as percentage of target cells cleareance) of total and sorted CD4+ and CD8+ T cells derived from three different healthy donors and redirected with e137-CARs (VH+VL and VL+VH) or S-CAR against HepG2 target cells stably transfected with HCV/E1–E2 glycoprotein of genotype 1a (H77 strain; A, C and D), transduced with human CD81 as control (E, G and H), or stably transfected with soluble HCV/E2 (HCV/sE2) of genotype 1a (H77 strain) (I). Untransduced total and sorted CD4+ and CD8+ T cells from the same donors were also used as control. Target cells were co-cultured in triplicate for 48 h with CAR-transduced T cells at four different effector to target (E:T) ratios, whereas untransduced T cells were used at a single 1:4 E:T ratio. Panels B, F and J showing the corresponding secreted interferon γ (IFN-γ) levels obtained from the different co-cultured total T cells against HepG2 target cells stably transfected with HCV/E1–E2, transduced with human CD81, or stably transfected with HCV/sE2, respectively. Levels of secreted HCV/sE2 (about 280 ng/mL of medium) were determined as described in the online supplementary methods. The mean plus SE from the mean (error bars) are reported. CAR, chimeric antigen receptor.
e137-CARs redirected T cells are activated by and lyse HCVcc-infected hepatocytes
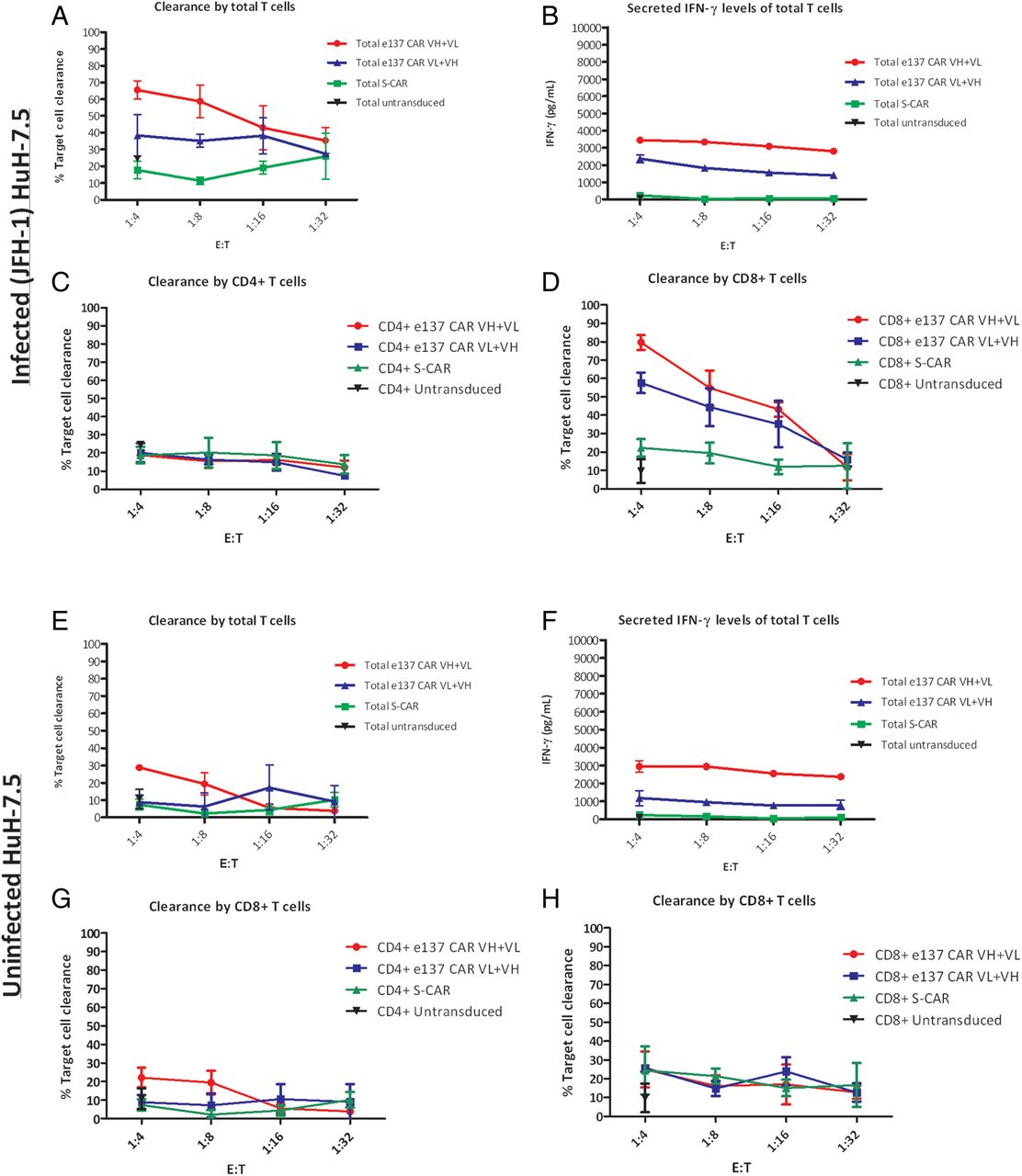
We assessed the cytotoxicity of redirected total and sorted CD4+/CD8+ T cells against HCVcc-infected (JFH-1 strain) HuH-7.5 cells. HCV/E2 antigen expression on the surface of infected cells was confirmed by fluorescence activated cell sorting (see online supplementary figure S5C). Attempts to infect HepG2 cells transduced with human CD81 gave very low efficiency of infection, as previously described elsewhere, and were hence not used in these experiments.43 e137-CARs-redirected T cells lysed HCVcc-infected HuH-7.5 cells (p<0.05 when compared to activity of S-CAR-redirected T cells), with an average of 65% and 40% of maximum target clearance for e137-CAR VH+VL-transduced and e137-CAR VL+VH-transduced total T cells at a 1:4 E:T ratio, respectively. A dose-dependent effect was observed using lower E:T ratios, more evidently for e137-CAR VH+VL-transduced T cells (figure 6A). Cytotoxicity of e137-CARs-redirected T cells was paralleled by significantly higher levels of proinflammatory cytokines and granzyme B compared to controls (p<0.01; figures 6B and online supplementary figure S9). However, in this experimental setting, a significantly higher rate of cytotoxicity (p<0.01) and even more notably, of IFN-γ secretion (p<0.01) were observed also on uninfected cells evidencing an aspecific cytotoxicity of e137-CARs-transduced T cells (especially in the VH+VL format) against HuH-7.5 cells (figure 6E–H). Interestingly, a higher average of 80% and 58% of maximum target clearance (1:4 E:T ratio) and a dose-dependent effect at lower E:T ratio were observed with e137-CAR VH+VL-transduced and e137-CAR VL+VH-transduced CD8+ T cells, respectively, showing also in this case specific target clearance compared to controls (p<0.05; figure 6D). Also in this case, cytotoxicity was accompanied by significantly higher proinflammatory cytokines and granzyme B levels compared to controls (p<0.01; see online supplementary figure S9).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cytotoxic activity of total and sorted CD4+ and CD8+ engineered T cells redirected with e137-CARs against infected and uninfected HuH-7.5 target cells. Panels showing the cytotoxic activity (expressed as percentage of target cells clearance) of total and sorted CD4+ and CD8+ T cells derived from three different healthy donors and redirected with e137-CARs (VH+VL and VL+VH) or S-CAR against cell culture-derived HCV (HCVcc)-infected (A, C and D) and uninfected (E, G and H) HuH-7.5 target cells. Untransduced total and sorted CD4+ and CD8+ T cells from the same donors were also used as control. Infected and uninfected target cells were co-cultured in triplicate for 48 h with CARs-transduced T cells at four different effector to target (E:T) ratios, whereas untransduced T cells were used at a single 1:4 E:T ratio. (B and F) The corresponding secreted interferon γ (IFN-γ) levels obtained from the different co-cultured total T cells against infected and uninfected HuH-7.5 target cells, respectively. The mean plus SE from the mean (error bars) are reported. CAR, chimeric antigen receptor.
Discussion
HCV is responsible for chronic liver disease worldwide and is associated with severe liver and systemic complications. The recent introduction of novel anti-HCV-specific antiviral drugs heralds important improvements in the treatment, recovery and life expectancy of infected patients.5 However, the lessons learned from other chronic infectious diseases, such as HIV and HBV, counsel us to be cautious, as possible resistances to direct-acting antivirals, occurring side effects and a consequent risk of a reduced compliance of the patient to the treatment regime could further contribute to the reduction of therapeutic efficacy.6 ,7 For these reasons, alternative strategies that could possibly be employed in selected categories of patients (ie, non-responders; end-stage patients undergoing liver transplantation) are certainly needed. In this regard, combining the cellular and humoral branches of the immune response in a single approach could represent a promising strategy. In this proof-of-concept study, we describe the engineering of human T cells through their transduction with human anti-HCV/E2 mAb-based CARs.
We constructed two anti-HCV/E2 CARs featuring two scFv variants of a previously described broadly cross-genotype reacting and cross-neutralising anti-HCV/E2 human mAb (e137).19 As preliminary steps, we evaluated the activation of anti-HCV/E2 CARs-redirected T cells and their capacity for clearing target cells expressing HCV/E1–E2 derived from different genotypes and subtypes. In this cell membrane-bound format of the antigen, the observed activity confirmed the accessibility of the highly conserved epitope recognised by the original mAb. Indeed, antigen recognition by T cells bearing e137-CARs resulted in degranulation and high-level secretion of granzyme B and proinflammatory cytokines IFN-γ, TNF-α and IL-2. Notably, engineered T cells bearing e137-CARs specifically cleared target cells transiently expressing the majority of the tested HCV/E1–E2 genotypes, including the more difficult-to-treat genotype 1. Overall, the cytotoxic activity exerted by the e137 scFv-engineered CARs against different HCV genotypes was not comparable to the binding and neutralising activities previously described for e137 as Fab fragment or as whole IgG1.19 ,21 As an example, e137-CARs were able to significantly clear cells expressing HCV/E1–E2 of genotypes 3a and 5, whereas the corresponding neutralising activity was lower than that observed against other genotypes, especially when using e137 as Fab.19 This is not surprising since it is well known that changes in the mAb format may markedly affect the binding of a given mAb to its antigen, as already observed also for e137.19 ,21 Moreover, it is important to bear in mind that the mechanisms of CAR activation are completely different from those associated to antibody-mediated viral neutralisation.33 In other words, the binding of a mAb to its epitope could not be sufficient to completely block the viral binding to its cellular receptors, thus resulting in an incomplete neutralisation, but, on the other hand, it could be sufficient to allow CAR clustering and T-cell activation.19 ,26 ,33
Considering that the hepatocytes are the main cellular target of HCV, cytotoxic assays were also performed on cell lines of hepatic origin, including stably transfected HepG2 with HCV/E1–E2 of genotype 1a (H77 strain), and a JFH1-derived (genotype 2a) HCVcc-infected HuH-7.5. In the same way as the results obtained with HEK cells, transfected HepG2 and infected HuH-7.5 cells were both lysed by e137-CARs-modified T cells, evidencing concomitant IFN-γ, IL-2, TNF-α and granzyme B secretion. In some experimental settings (ie, e137 CAR VH+VL-redirected T cells against stably HCV/E1–E2-transfected HepG2), the levels of secreted cytokines did not directly correlate with the cytotoxic activity of T cells. This became more evident when using total T cells, and it is probably due to the prevalent expression of proinflammatory cytokines instead of cytotoxic-related proteins (eg, LAMP-1 and granzyme B), following antigen recognition and CD28 costimulatory signal. This could be further due to the general lower level of CD8+ compared to CD4+ T cells, as observed in the original unsorted population used in our assays (see online supplementary figure S2). In fact, the cytotoxic activity markedly increased when only CD8+ T cells were sorted and co-cultured with HCV/E1–E2 stably transfected target cells, as reported in other studies on animal models and clinical trials employing CAR-redirected T cells only belonging to the CD8+ subset.40 ,44 ,45
Conversely, the IFN-γ levels stimulated by HCVcc-infected HuH-7.5 were only moderately increased if compared to those observed with HCV/E1–E2-transfected target cells. Moreover, a higher level of aspecific cytotoxic activity against uninfected HuH-7.5 cells was also observed. This is in accordance with previously published data indicating that HuH-7 as well as HuH-7.5 are particularly susceptible to human T cells, and that expression of full HCV genome induces low IFN-γ production by native CD4+ T cells.32 ,46 Furthermore, background activity of CAR-grafted T cells against hepatocyte-derived target cell lines has been previously observed.40 As far as IFN-γ is concerned, the explanation for the differing results noted between the two experimental settings using hepatic cell lines could be because the surface-expressed antigen is more abundant on transfected than on infected target cells.12 However, in the case of infected HuH-7.5 cells, a more specific cytotoxicity and secretion of proinflammatory cytokines was also observed when using sorted CD8+ e137-CARs-transduced T cells.
Finally, as demonstrated in experiments employing target cells (HepG2 and HEK-293) expressing a secreted form of HCV/E2 (sE2), the presence of soluble antigen in the medium, as well as its transitory and minimal expression at the level of the cell surface during the trafficking of the protein,12 ,47 is not able to trigger a significant major activation of e137-CARs-transduced T cells. Data were also confirmed by directly culturing e137-CARs-grafted T cells in presence of HCV/sE2 or HCVcc viral-particles. This may have important implications for the possible use of e137-based CARs in vivo. It has been demonstrated that HCV is usually present at high titre in the bloodstream of infected patients. Indeed, circulating viral particles could interfere or block the activity mediated by the CAR, as well as induce its extrahepatic activation.48 From a molecular point of view, we speculate that e137-CARs-grafted T cells are able to be activated by full-length HCV/E2, mostly expressed as a cluster at the level of the target cell membrane (a condition also simulated in the case of a plate-coated antigen), thus leading to proper activation of CAR-associated intracellular signalling machinery, and to the degranulation and secretion of granzyme B, and proinflammatory cytokines. The lack of interference by free antigen was not obvious, since one of the few papers previously describing anti-infective CARs, reported an interfering effect of free HIV/gp120, another hypervariable envelope glycoprotein, on the lytic activity of an anti-HIV/gp120 CAR.49 In our case, the conserved nature and the structural importance of the epitope recognised by the original mAb on HCV/E2 probably favoured proper CAR-clustering.49
Although the activity of redirected T cells is very promising, the safety profile of engineered T cells must be carefully considered before any possible clinical use. The translation of CAR-engineered T cells into clinics has been reported in several studies mostly focused on oncological diseases. For example, a recent clinical trial reported for the first time a complete response 3 weeks after treatment with autologous T cells genetically modified to target CD19 in a chronic lymphoid leukaemia-affected patient.50 Several studies have also addressed the potential risk of CAR-engineered T cells extensive proliferation and cytotoxicity in vivo by the introduction of suicide genes in the transducing virus, in order to selectively eliminate modified T cells after remission or in case of aberrant proliferation.33 In this regard, Bohne et al51 and Krebs et al40 recently described the use of anti-HBV CAR-transduced T cells in vitro as well as against HBV-replicating hepatocytes of immune competent transgenic mice expressing the HBV genome. The anti-HBsAg S-CAR was able to reduce HBV replication in mice, causing only temporary liver damage. Interestingly, circulating HBV antigens did not weaken or over-activate S-CAR-grafted T cells, a feature shared by the anti-HCV/E2 CARs described in this paper.40 Different surrogate murine models have been engineered to support HCV infection in order to circumvent the lack of a small animal model for this infection. However, at present, none of these represent a valid and robust in vivo model. In fact, as in the case of those available for HBV, all of these are burdened by several limitations, such as high variability of viral replication, short life expectancy and low overall robustness, factors rendering the interpretation of data extremely difficult.52 In this respect, uPA-SCID mice engrafted with human hepatocytes, represented a significant milestone in the development of a small animal model for HCV infection. However, the above limitations, as well as the obtained low titre of infected hepatocytes, restrict the use of this model. Transgenic mice expressing HCV proteins have also been described, in particular for the possibility of their role in the study of HCV in the liver pathogenesis. However, these proteins are expressed as self-antigens making difficult a clear evaluation of the antiviral effect of possible molecules or engineered T cells. On the other hand, transgenic mouse models expressing human entry factors for HCV infection have also been described. However, viral replication is not efficiently supported in the cells of these animal models, complicating the interpretation of the possible efficacy of the candidate antiviral agent.53 At the moment, chimpanzees represent the only available animal model, but other limits, including ethical and economic consideration issues, restrict their use.52
In this proof-of-concept study, we made a preliminary investigation into the possible antiviral in vitro activity of CAR-engineered T cells redirected against HCV/E2 glycoprotein expressed on the surface of transfected and infected cells. Thanks to recent advances in stem cell and tissue engineering technologies, the gradual improvement in existing mouse models for HCV infection could allow future in vivo studies to better determine the effectiveness and safety profile of this novel antiviral strategy before a possible translation into clinics. With regard to this, promising immunocompetent mouse models engrafted with human hepatocytes and with human immune cell progenitors have very recently been developed.54 ,55
In conclusion, in this study we have constructed two second generation CARs targeting HCV/E2 glycoprotein and retrovirally delivered it to primary human T cells. The two CARs featured the binding characteristics of a previously described cross-reactive and cross-neutralising mAb (e137), and showed a similar activity, since e137-CARs-modified T cells were activated, and lysed HCV/E2-expressing cells and HCVcc-infected hepatocytes. A possible future combination therapy in patients experiencing treatment failure with available molecules, or in those at risk of re-infection, such as patients undergoing liver transplantation, could include broadly cross-neutralising anti-HCV/E2 mAbs and infusion of anti-HCV/E2 CAR-modified autologous or HLA-compatible T cells, such as e137 mAb and CARs, targeting highly conserved regions of the antigen and crucial for the viral life-cycle.19 This novel approach targeting a broadly conserved and functionally pivotal HCV/E2 epitope could have a direct effect on at least two of the most important HCV escape mechanisms from the humoral and the cellular immune response, such as viral hypervariability and cell-to-cell transmission. As a consequence, a marked impairment of HCV replicative fitness could be observed which, even if not completely eradicating the infection, could markedly limit its pathogenic sequelae.
Acknowledgments
The authors would like to thank Dr Birke Bartosch (Centre de Recherche en Cancérologie de Lyon, France) for providing the plasmid and the retroviral supernatants for transduction of HepG2 cells with human CD81, Dr Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan) for providing the construct to generate HCVcc (JFH-1 strain), Prof Hinrich Abken (University Hospital Cologne, Germany) for providing plasmid containing CD28 and CD3 signalling domains, Dr Alexander W Tarr (Queen's Medical Centre, University of Nottingham, UK) for providing plasmids encoding the different HCV/E1–E2 isolates and mutants, Dr Giacomo Gorini and Theresa Asen for valuable technical assistance. The authors also thank Dan McAuley for revising the English manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
Contributors GAS and NM designed and conceived the study and wrote the manuscript. GAS and KW acquired, analysed and interpreted the data. MCa, RAD and JG technically supported the study. GAS and NC performed the statistical analysis. KW, NC, MCa, MCl, RB and UP critically revised the manuscript. NM, MCl, RB and UP supervised the study.
Funding This study was partially supported by the German research foundation (DFG), CRC_TR 36.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.