

Abstract
Regulated expression and secretion of BDNF, which activates TrkB receptor signaling, is known to play a critical role in cognition. Identification of additional modulators of cognitive behavior that regulate activity-dependent BDNF secretion and/or potentiate TrkB receptor signaling would therefore be of considerable interest. In this study, we show in the adult mouse hippocampus that expression of the granin family gene Vgf and secretion of its C-terminal VGF-derived peptide TLQP-62 are required for fear memory formation. We found that hippocampal VGF expression and TLQP-62 levels were transiently induced after fear memory training and that sequestering secreted TLQP-62 peptide in the hippocampus immediately after training impaired memory formation. Reduced VGF expression was found to impair learning-evoked Rac1 induction and phosphorylation of the synaptic plasticity markers cofilin and synapsin in the adult mouse hippocampus. Moreover, TLQP-62 induced acute, transient activation of the TrkB receptor and subsequent CREB phosphorylation in hippocampal slice preparations and its administration immediately after training enhanced long-term memory formation. A critical role of BDNF-TrkB signaling as a downstream effector in VGF/TLQP-62-mediated memory consolidation was further revealed by posttraining activation of BDNF-TrkB signaling, which rescued impaired fear memory resulting from hippocampal administration of anti-VGF antibodies or germline VGF ablation in mice. We propose that VGF is a critical component of a positive BDNF-TrkB regulatory loop and, upon its induced expression by memory training, the TLQP-62 peptide rapidly reinforces BDNF-TrkB signaling, regulating hippocampal memory consolidation.
SIGNIFICANCE STATEMENT Identification of the cellular and molecular mechanisms that regulate long-term memory formation and storage may provide alternative treatment modalities for degenerative and neuropsychiatric memory disorders. The neurotrophin BDNF plays a prominent role in cognitive function, and rapidly and robustly induces expression of VGF, a secreted neuronal peptide precursor. VGF knock-out mice have impaired fear and spatial memory. Our study shows that VGF and VGF-derived peptide TLQP-62 are transiently induced after fear memory training, leading to increased BDNF/TrkB signaling, and that sequestration of hippocampal TLQP-62 immediately after training impairs memory formation. We propose that TLQP-62 is a critical component of a positive regulatory loop that is induced by memory training, rapidly reinforces BDNF-TrkB signaling, and is required for hippocampal memory consolidation.
Introduction
Neurotrophins, including BDNF, are stored in large dense-core vesicles (LDCVs) and are secreted in response to increased neuronal activity, which in turn regulates synaptic plasticity and the strength of circuit connectivity (Blum and Konnerth, 2005; Park and Poo, 2013), contributing to memory formation (Tyler et al., 2002; Minichiello, 2009; Bekinschtein et al., 2014; Deinhardt and Chao, 2014; Lynch et al., 2014). Upstream modulators of regulated BDNF secretion and subsequent TrkB receptor activation are still largely unknown, although aberrant BDNF sorting and regulated secretion resulting from the Val/Met BDNF polymorphism is a recognized contributor to neuropsychiatric disease (Egan et al., 2003; Chen et al., 2006).
VGF (nonacronymic) is a BDNF-inducible peptide precursor and secreted granin protein (Bartolomucci et al., 2011; Ferri et al., 2011). In the CNS, VGF-derived peptides regulate neuronal activity (Alder et al., 2003; Bozdagi et al., 2008), neuronal survival (Zhao et al., 2008; Sato et al., 2012), and neural progenitor proliferation (Thakker-Varia et al., 2007). The C-terminal VGF-derived peptide TLQP-62 (named by the four N-terminal amino acid residues and length) enhances neuronal transmission through a BDNF-TrkB-dependent pathway in hippocampal slices (Bozdagi et al., 2008). In addition, full-length VGF1–617 functions in the regulated secretory pathway, modulating formation of LDCVs (Fargali et al., 2014).
The expression of VGF in neurons is robustly regulated by neurotrophic growth factors (Salton et al., 1991; Alder et al., 2003) and by neuronal activity (Snyder et al., 1998b). VGF is widely expressed in the developing and adult brain, including in cerebral cortex, hypothalamus, and hippocampus (van den Pol et al., 1994; Lombardo et al., 1995; Snyder and Salton, 1998; Snyder et al., 1998a). Decreased VGF levels in CSF, hippocampus, or prefrontal cortex have been reported in patients with Alzheimer's disease, frontotemporal dementia, and psychiatric disorders including bipolar disorder, schizophrenia, and social anhedonia (Rüetschi et al., 2005; Simonsen et al., 2008; Thakker-Varia et al., 2010; Ramos et al., 2014). Studies in germline-targeted VGF knock-out mice have also identified a role(s) for VGF in memory formation (Bozdagi et al., 2008) and stress-induced depression (Hunsberger et al., 2007; Thakker-Varia et al., 2007). However, the underlying mechanisms by which VGF and VGF-derived peptides regulate memory formation, and the regions of the adult brain that are involved, are still largely unknown.
Here, we investigated the roles that VGF and the VGF-derived C-terminal peptide TLQP-62 play in long-term memory formation, by conditionally ablating the expression of VGF or blocking the function of secreted VGF peptides in adult dorsal hippocampus. The mechanism by which VGF and TLQP-62 mediate fear memory formation involves the activation of BDNF-TrkB signaling pathways and subsequent neuronal cytoskeletal reorganization, potentially affecting synaptic structure and function (Hale et al., 2011; Deinhardt and Chao, 2014; Lynch et al., 2014). Our data suggest that adult VGF expression is required to potentiate BDNF-TrkB receptor signaling, which contributes critically to a positive autoregulatory feedback loop that mediates hippocampal-dependent memory consolidation (Canossa et al., 1997; Cheng et al., 2011; Bambah-Mukku et al., 2014).
Materials and Methods
Generation of Vgf germline and floxed mouse lines.
The floxed VGF mouse line was generated by inserting a 5′ flanking loxP site into the Vgf 5′ UTR (KpnI site), and a 3′ flanking loxp site and inverted FRT-flanked neomycin selection cassette derived from p-loxP-2FRT-PGKneo (Dr. David Gordon, University of Colorado Health Science Center) into the Vgf 3′ UTR (XbaI site) using previously described mouse Vgf genomic sequences (Hahm et al., 1999). The construct was electroporated into hybrid 129B6 ES cells (inGenious Targeting Laboratory, Stony Brook, NY), and male chimeras were mated with C57BL/6J females to produce F1 breeders having germline transmission of the targeted, floxed Vgf allele. Experiments were performed on male mixed background N3F1 Vgfflox/flox (>93.7% C57BL/6J background) and wild-type littermates. Vgfflpflox/flpflox mice were generated by crossing Vgfflox/flox with B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/JRainJ (JAX: 009086; The Jackson Laboratory) mice to remove the FRT-flanked neomycin cassette in the germline, after which these mice were backcrossed to C57BL/6J. Vgfflpflox/flpflox mice (>98% C57BL/6J background; MAX-BAX; Charles River Laboratories) were then crossbred with αCaMKII-Cre transgenic mice (JAX: 005359; The Jackson Laboratory) to generate forebrain VGF knock-out mice. Experiments were performed on male N5F1 αCaMKII-Cre/+; Vgfflpflox/flpflox mice and αCaMKII-Cre/+; Vgf+/+ littermates. The VGF germline knock-out mouse line was generated as described previously (Fargali et al., 2014) and experiments were performed on male N2F1 VGF heterozygous knock-out mice and wild-type littermates. Animals were housed on a 12 h light–dark cycle with ad libitum access to food and water. All animal studies were conducted in accordance with the National Institutes of Health's Guidelines for the Care and Use of Experimental Animals using protocols approved by the Institutional Animal Care and Use Committee of the Icahn School of Medicine at Mount Sinai and New York University.
Stereotaxic injections and cannula implants.
Male C57BL/6J mice (2–3 months old; Charles River Laboratories) were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg). Thirty-three gauge syringe needles (Hamilton) were used to bilaterally infuse 1.0 μl of AAV virus or colchicine (100 μg) into mouse dorsal hippocampus [AP = −2.0, ML= ±1.5, and DV = −2.0 from bregma (mm); Kaspar et al., 2002] at a rate of 0.2 μl per min and the needle remained in place for 5 min before removal to prevent backflow. AAV-CreGFP and AAV-GFP (AAV2 vector, serotype AAV5) were purchased from the Vector Core at the University of North Carolina at Chapel Hill. AAV-injected mice were allowed to recover for 28 d before behavioral testing. For antibody infusion, different cohorts of mice were used for each experiment and a double-guide cannula (22-gauge) was implanted to target dorsal hippocampus bilaterally. Mice were allowed to recover for 7 d before behavioral testing.
Long–Evans adult male rats (Harlan Laboratories) weighing between 200 and 250 g at the beginning of the experiments were used. Rats were anesthetized with ketamine (65 mg/kg) and xylazine (7.5 mg/kg) and guide cannulae (22-gauge) were stereotactically implanted to target the dorsal hippocampus bilaterally [AP = −4.0, ML = ±2.6, and DV = −3.5 from bregma (mm)]. The rats were returned to their home cages and allowed to recover for 7 d before behavioral testing.
Antibodies and peptide infusion.
Different cohorts of mice were used for each experiment. Immediately after memory training (contextual fear conditioning for mice and inhibitory avoidance for rats), animals received bilateral injections of antibodies or peptides as specified. All hippocampal injections (1 μl per side) were performed using a 28-gauge internal cannula at a rate of 0.2 μl per min (mouse) or 0.33 μl per min (rat) using a microinfusion pump (Harvard Apparatus). The injection needle was left in place for 2 min after the injection to allow complete dispersion of the solution. All animals were awake and were allowed to move freely throughout the infusion. Antibodies and peptides used for infusion were as follows: anti-VGF C-terminal antibody (anti-VGF565–615 IgG; Santa Cruz Biotechnology), anti-AQEE-30 and anti-TLQP-21 IgG (Fairbanks et al., 2014), synthetic peptides TLQP-62 and its scrambled control peptide SC-62 (GenScript), and BDNF (PeproTech).
Contextual fear conditioning.
Male mice (2- to 3-month old for C57BL/6J or VGF germline knock-out, and 3∼5-month old for αCaMKII-Cre/+; Vgfflpflox/flpflox) were used for contextual fear conditioning (CFC) studies. Fear conditioning took place in an observation chamber (30 × 24 × 21 cm; MED Associates). All experiments were performed during the light cycle between 9:00 A.M. and 6:00 P.M. Mice were handled in the room where conditioning took place for 3 min each day for 5 d. On the training day, they were placed into the chamber and, after a 3 min acclimatizing period, they received two shocks (0.75 mA for 2 s) at 60 s intervals for strong training or one shock (0.3 mA for 2 s) for weak training. Mice were left in the conditioning chamber for 30 s after termination of the procedure and then returned to their home cages. For the rescue experiment, the TrkB agonist 7,8-dihydroxyflavone (7,8-DHF; dissolved in 17% DMSO in PBS; Sigma-Aldrich) was administered by intraperitoneal injection (5 mg/kg) immediately after training or antibody infusion, as indicated. One hour or 24 h later, mice were placed back into the conditioning chamber and observed for 3 min. Freezing behavior was recorded blind to genotype or the treatments given during acclimatizing and testing sessions. Freezing was defined as the absence of any visible movements other than those necessary for respiration and was scored according to a 10 s time sampling procedure. Observations scored as freezing were summed and converted to a percentage. For locomotor activity, a video-tracking system (Ethovision 3.0; Noldus) was used to measure the total distance traveled (in centimeters) in the arena (open field arena, 44 × 44 cm) for 10 min.
Inhibitory avoidance.
Male rats were housed individually and all inhibitory avoidance (IA) experiments were performed during the light cycle between 9:00 A.M. and 6:00 P.M., as described previously (Chen et al., 2012). All rats were handled for 3 min per day for 5 d before any behavioral procedure or memory testing. During training sessions, each rat was placed in the safe compartment of the two-chamber IA box (MED Associates) with its head facing away from the door. The door closed 1 s after the rat entered the shock compartment and a brief foot shock (0.9 mA for 2 s) was administered. Latency to enter the shock compartment was taken in seconds as acquisition. The rat was then returned to its home cage and tested for memory retention at the designated time points (2 and 7 d after training). Retention tests were done by placing the rat back in the safe compartment and measuring its latency to enter the shock compartment. During testing, the experimenter was blind to the treatments given.
Western blot and dot-blot analysis.
Mouse dorsal hippocampal extracts were obtained by tissue homogenization in ice-cold RIPA lysis buffer (1× PBS buffer containing 1% NP-40, 0.5% Na deoxycholate, and 0.1% SDS) with a protease inhibitor mixture (Roche) and a phosphatase inhibitor mixture (Sigma-Aldrich). Protein concentrations were determined using the Bio-Rad protein assay. Equal amounts of total protein (10 μg per lane) were resolved on denaturing SDS-PAGE gels and transferred to Hybond-P membranes (Millipore) by electroblotting. The membranes were then blocked in 10% milk/PBS for 1 h at room temperature and then incubated with anti-VGF C-terminal (1/1000, rabbit polyclonal), anti-phospho-CREB (1/1000; Cell Signaling Technology), anti-CREB (1/1000; Cell Signaling Technology), anti-phospho-TrkB (1/5000; Epitomics), anti-TrkB (1/1000; Abcam), anti-phospho-cofilin (1:1000; Abcam), anti-cofilin (1:1000; Cell Signaling Technology), anti-phospho-synapsin-1 (1:1000; Sigma-Aldrich), anti-BDNF (1:500; Santa Cruz Biotechnology), or anti-actin (1/1000; Sigma-Aldrich) antibodies in 1× PBS (3% BSA) overnight at 4°C. The membranes were washed, incubated with a secondary horseradish peroxidase-labeled donkey anti-rabbit or donkey anti-mouse antibody (1/6000; GE Healthcare) for 1 h, washed again, and incubated with ECL detection reagents (Millipore). Densitometric analysis was performed using ImageJ software. Dot-blot analysis was performed as described previously (Fairbanks et al., 2014). Briefly, peptides were blotted on PVDF-FL membranes and incubated in blocking buffer (Odyssey; LI-COR Biosciences) for 1 h, followed by primary antibody incubation overnight at 4°C, secondary antibody incubation for 1 h (IRDye800 goat anti-rabbit, IRDye800 goat anti-guinea pig; LI-COR Biosciences) and imaging (Odyssey Imaging System; LI-COR Biosciences).
Immunohistochemistry.
Mice were anesthetized with a ketamine/xylazine mixture and intracardially perfused with ice-cold 4% paraformaldehyde (in 1× PBS). Brains were postfixed for 4 h at 4°C before vibratome sectioning (50 μm; Leica VT 1000S). Free-floating sections were permeabilized with 0.2% Triton X-100 in PBS at room temperature and then blocked in 3% goat serum and 5% bovine serum albumin (BSA) with 0.05% Triton X-100 in PBS for 1 h. Brain sections were incubated in 1:2000 anti-VGF C-terminal (rabbit polyclonal Ab or guinea pig polyclonal Ab), 1:2000 anti-GABA (Sigma-Aldrich), or 1:1 anti-GAD-6 (Developmental Studies Hybridoma Bank, University of Iowa) in 5% BSA overnight at 4°C. The next day, sections were washed with 0.2% Triton X-100 in PBS and then incubated in 1:500 goat anti-rabbit Texas Red X (Life Technologies) or 1:500 goat anti-guinea pig Alexa Fluor 488 (Life Technologies) in PBS for 1 h and then washed again. All sections were stained with DAPI, mounted with antifade solution (Life Technologies), and imaged with a Zeiss Imager Axio M1 or Zeiss LSM780 confocal microscopes. The pixel intensity of VGF was quantified using ImageJ.
RNA extraction and qPCR analysis.
Total RNA was extracted by using TRIzol (Life Technologies) according to the manufacturer's protocol. Total RNA (0.5 μg) was reverse transcribed using qScript cDNA synthesis kit (Quanta Biosciences). One nanogram of the first-strand cDNA product was subjected to PCR amplification using a SYBR green real-time reverse transcription PCR (qPCR) master mix (PerfeCta SYBR Green FastMix; Quanta Biosciences). Amplification was performed on an ABI Prism 8500 system (Mount Sinai Shared Resource Facility) in triplicate. SDS 2.1 software was used for analyzing cycle threshold (Ct) values. The Ct method (see Applied Biosystems User Bulletin Number 2, P/N 4303859) and ΔΔCt were used to determine the relative quantification of gene expression in trained and naive mice. Primers for mouse Rac1-forward, 5′-GGTAGGTGATGGGAGTCAGC-3′, mouse Rac1-reverse, 5′-CTGAAGTGCGACACCACTGT-3′, mouse c-fos-forward, 5′-CCGAAGGGAACGGAATAAGA-3′, mouse c-fos-reverse, 5′-TGCAACGCAGACTTCTCATCT-3′, mouse Vgf-forward, 5′-GGTAGCTGAGGACGCAGTGT-3′, mouse Vgf-reverse, 5′-GTCCAGTGCCTGCAACAGTA-3′, mouse Gapdh-forward, 5′-GAACATCATCCCTGCATCCA-3′, mouse Gapdh-reverse, 5′-CCAGTGAGCTTCCCGTTCA-3′.
Hippocampal slice preparation and treatment.
Hippocampal slices (350 μm) were prepared from 2- to 3-month old C57BL/6J mice as described previously (Bozdagi et al., 2008). Slices were perfused with Ringer's solution containing the following (in mm): 125.0 NaCl, 2.5 KCl, 1.3 MgSO4, 1.0 NaH2PO4, 26.2 NaHCO3, 2.5 CaCl2, and 11.0 glucose. The Ringer's solution was bubbled with 95% O2/5% CO2 at 32°C during peptide treatment. Slices were maintained for 1 h before treatment with synthetic VGF peptides TLQP-62 (10 μm, C-terminal 62 aa rat VGF-derived peptide) or its scrambled peptide control SC-62 (10 μm). For blocking, slices were preincubated with TrkB-Fc scavenger (5 μg/ml) for 30 min before peptide treatment. Slices were collected at indicated time points (10 min or 30 min) and immediately frozen on dry ice.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism 5 software. Comparisons were made using two-tailed Student's t test, one-way or two-way ANOVA for repeated measures, followed by Tukey or Bonferroni post hoc tests. The p-values and specific tests used are indicated in the figure legends or the Results section.
Results
CFC increases VGF expression acutely in brain areas critical for memory formation
Learning deficits previously identified in homozygous and heterozygous VGF germline knock-out mice could reflect a requirement for VGF during brain development and/or in critical adult brain circuits that are required for memory acquisition and/or consolidation (Bozdagi et al., 2008). To examine directly the functional role of VGF in the adult brain, we used two approaches. First, we examined whether VGF expression in the dorsal hippocampus, a brain region critical for CFC memory consolidation, is induced after fear memory training. Second, to bypass the potential critical contribution(s) of VGF during development, we generated VGF conditional knock-out mouse lines that allow us to manipulate VGF expression in selected regions and neuronal types in the adult brain.
We found that VGF expression, both mRNA and protein, was increased 1 h after training in the dorsal hippocampus (Fig. 1a). Vgf mRNA levels peaked 1 h after training, whereas the levels of both VGF protein and its processed C-terminal peptide TLQP-62 progressively increased 1 and 6 h after training. As a positive control, we examined expression of c-fos, an immediate early gene critical for memory formation that is acutely induced after memory training (Huff et al., 2006). Indeed, increased c-fos mRNA levels were also detected 1 h after training (Fig. 1a). In addition, previous studies have shown that BDNF signaling is required for hippocampal-dependent memory consolidation through activation of TrkB receptor (auto-phosphorylation; Alonso et al., 2002b; Liu et al., 2004); we also found an increase in phospho-TrkB levels in the hippocampus 1 h after training (Fig. 1a). Notably, training did not increase VGF protein levels in the dorsal hippocampus of VGF germline heterozygous knock-out mice (Fig. 1b), which have impaired contextual fear memory (Bozdagi et al., 2008), suggesting that the levels of basal and inducible VGF expression may both be critical for hippocampal-dependent memory formation because they play a related role in depressive behavior and the antidepressant response to exercise (Hunsberger et al., 2007).

Fear memory training increases VGF expression in adult mouse hippocampus. a, Quantification of VGF protein and mRNA expression in mouse dorsal hippocampus after contextual fear conditioning. A significant increase in both VGF protein and its processed C-terminal TLQP-62 peptide levels was observed 1 and 6 h after training (VGF full-length protein: ANOVA, F(3,26) = 26.27, p < 0.0001; TLQP-62 peptide: ANOVA, F(3,25) = 4.915, p = 0.0081). TrkB receptor phosphorylation was also induced 1 h after training (ANOVA, F(3,20) = 5.449, p = 0.0066). Similar to c-fos, Vgf mRNA levels peaked 1 h after training. Vgf (RNA): ANOVA, F(3,16) = 18.65, p < 0.0001; c-fos (RNA): ANOVA, F(3,16) = 11.74, p = 0.0003. N = 4–12 (protein), n = 3–8 (RNA). One-way ANOVA followed by Tukey's post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001. b, VGF protein levels fail to increase 1 and 6 h after CFC training in the dorsal hippocampus of VGF heterozygous knockout mice (VGF protein: ANOVA, F(5,20) = 9.30, p = 0.0001). N = 3–5. One-way ANOVA followed by Tukey's post hoc test. *p < 0.05, **p < 0.01. c, Brain sections were prepared from naive or 6 h memory-trained mice and stained with rabbit anti-VGF (C-terminal) polyclonal antibody. Increased VGF staining was observed in the pyramidal layer of CA1, CA2/3, and hilus region. n = 3 per group. Data were analyzed by Student's t test. *p < 0.05. Scale bar, 50 μm.
To further confirm and extend our findings, we applied immunohistochemical staining with a specific rabbit polyclonal antibody raised against the C-terminal region of the VGF protein. Six hours after fear memory training, increased VGF protein levels were detected in all subregions of dorsal hippocampus (p < 0.05, Student's t test; Fig. 1c), suggesting that, in the adult hippocampus, induction of VGF protein and its C-terminal-derived TLQP-62 peptide throughout the dorsal hippocampus is associated with fear memory formation.
Downregulation of hippocampal VGF expression in adult mice disrupts memory retention
To generate conditional VGF knock-out mouse lines, the Vgf locus in mouse embryonic stem cells was targeted and flanking loxP sites and an inverted FRT-flanked neomycin selection cassette were inserted (Vgfflox/flox line; Fig. 2a). To determine whether adult hippocampal VGF expression is important for memory formation, AAV-CreGFP virus, which encodes a Cre-GFP fusion protein, was injected into the dorsal hippocampus of adult Vgfflox/flox mice. Significantly decreased VGF immunohistochemical staining was observed in the CA2/3 and hilus regions compared with AAV-GFP virus-injected controls (44.1% decrease in CA2/3 region, p < 0.01; 41.5% decrease in hilus region, p < 0.05, Student's t test; Fig. 2b). Ablation of VGF in adult dorsal hippocampus resulted in a long-term memory deficit (p < 0.01; Fig. 2c), but no significant differences in short-term memory or locomotor activity (Fig. 2d,e). We also investigated whether VGF knock-down in the adult hippocampus of naive animals affected the level or phosphorylation of several proteins known to be associated with memory consolidation. We found that AAV-CreGFP injection reduced VGF protein levels, but did not significantly affect pCREB, total CREB, pTrkB, total TrkB, or BDNF levels in the hippocampus (28 d after AAV injection into Vgfflpflox/flpflox mice; Fig. 2f). Collectively, our results suggest that VGF expression in the adult hippocampus is required for the consolidation of, but not the acquisition of, contextual fear memory.

Downregulation of VGF expression in adult mouse hippocampus impairs long-term memory formation. a, Gene-targeting strategy for deleting mouse Vgf alleles. Two loxP sites (red arrowhead) were inserted into the second exon and 3′UTR of the Vgf gene, flanking the entire VGF coding sequence (Vgfflox/flox mice) and allowing its removal by AAV-CreGFP-mediated recombination. In experiments using αCaMKII-Cre-mediated VGF deletion, we used mice that had the inverted neomycin selection cassette, flanked by two FRT sites (yellow arrowheads), removed by FLP recombinase-mediated deletion (Vgfflpflox/flpflox mice). b, Four weeks after AAV-CreGFP virus injection into the dorsal hippocampus of Vgfflox/flox mice, immunohistochemical staining showed reduced VGF levels in CA2/3 region and hilus (red: rabbit anti-VGF (C-terminal) polyclonal Ab; green: GFP). n = 3 per group. Fluorescence intensity was analyzed by Student's t test. *p < 0.05, **p < 0.01. Scale bar, 100 μm. c, AAV-CreGFP injected Vgfflox/flox adult mice showed decreased freezing 24 h after memory training (n = 11∼14 per group, two-way ANOVA, main effect of virus, F(2,72) = 9.76, p = 0.0002; main effect of training, F(1,72) = 446.73, p < 0.0001; Bonferroni post hoc test. ***p < 0.001). d, e, No significant differences were found in short-term memory (n = 6∼7 per group, one-way ANOVA, F(2,17) = 1.75, p = 0.20) and locomotor activity (n = 6∼7 per group, one-way ANOVA, F(2,17) = 0.36, p = 0.69) compared with AAV-GFP-injected wild-type (Vgf+/+) or Vgfflox/flox controls. Bars represent average freezing (%) or total distance traveled (cm) ± SE. f, AAV-CreGFP injection reduced VGF protein levels, but did not significantly affect protein levels of hippocampal pCREB, total CREB, pTrkB, total TrkB, and BDNF levels. n = 6∼7 per group, value represents average protein level of AAV-GFP (%) ± SE, Student's t test, ***p < 0.001.
VGF ablation in adult forebrain excitatory neurons disrupts memory
VGF protein expression has been reported previously in hippocampal mossy fiber terminals and interneurons of rodent brain (van den Pol et al., 1994; Hunsberger et al., 2007). To facilitate detection of cells that express the secreted protein VGF (van den Pol et al., 1994), we administered colchicine, which inhibits microtubule polymerization and blocks neuropeptide secretion, into dorsal hippocampus 24 h before immunohistochemical staining. Colchicine treatment resulted in robust VGF staining in the pyramidal layer of CA1–3, stratum oriens of CA1–3, stratum radiatum of CA3, stratum lacunosum-moleculare of CA1–3, and hilus region, but only scattered staining in the granular layer of dentate gyrus (Fig. 3a), in agreement with the previously reported pattern of VGF mRNA expression in the adult dorsal hippocampus revealed by in situ hybridization (Snyder and Salton, 1998). VGF-expressing cells in the stratum oriens, stratum radiatum, stratum lacunosum-moleculare, and hilus regions also coexpress GABA, indicating that VGF can be expressed by both excitatory and inhibitory neurons in the hippocampus (Fig. 3b).

Enhanced VGF protein staining detected in both excitatory and GABAergic inhibitory neurons in the hippocampus. a, A robust increase of VGF signal in cell bodies was detected in mouse dorsal hippocampus 24 h after colchicine injection (right) compared with un-injected brain (left). Scale bar, 100 μm. b, Double-labeled immunohistochemical staining of colchicine-injected hippocampus showed VGF labeling in the cell bodies of both GABAergic and pyramidal neurons in all regions of dorsal hippocampus [red, anti-GABA Ab; green, guinea pig anti-VGF (C-terminal) polyclonal Ab]. Scale bar, 100 μm.
To further examine the functional role of VGF in excitatory neurons in the neocortex and hippocampus, we generated conditional αCaMKII-Cre/+;Vgfflpflox/flpflox mice made by removing the FRT-flanked neomycin cassette from Vgfflox/flox mice through crossbreeding with FLP-recombinase expressing B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym mice; the resulting Vgfflpflox/flpflox were crossbred with αCaMKII-Cre transgenic mice (Fig. 2a). Cre expression driven by the αCaMKII promoter in this line has been shown previously to be restricted to adult postnatal excitatory forebrain neurons (Tsien et al., 1996). Immunohistochemical staining results from αCaMKII-Cre/+;Vgfflpflox/flpflox mice showed VGF downregulation in the CA1 and CA3 pyramidal neurons of adult hippocampus, whereas VGF levels in GAD-65-positive interneurons remained unchanged compared with αCaMKII-Cre/+;Vgf+/+ littermates (Fig. 4a). Interestingly, forebrain VGF ablation specifically in postnatal excitatory neurons also resulted in impaired long-term memory formation (p < 0.01; Fig. 4b), whereas short-term memory and locomotor activity were not affected (Fig. 4b,c). Our results, therefore, indicate a critical role for VGF synthesized in excitatory adult forebrain neurons in memory consolidation.
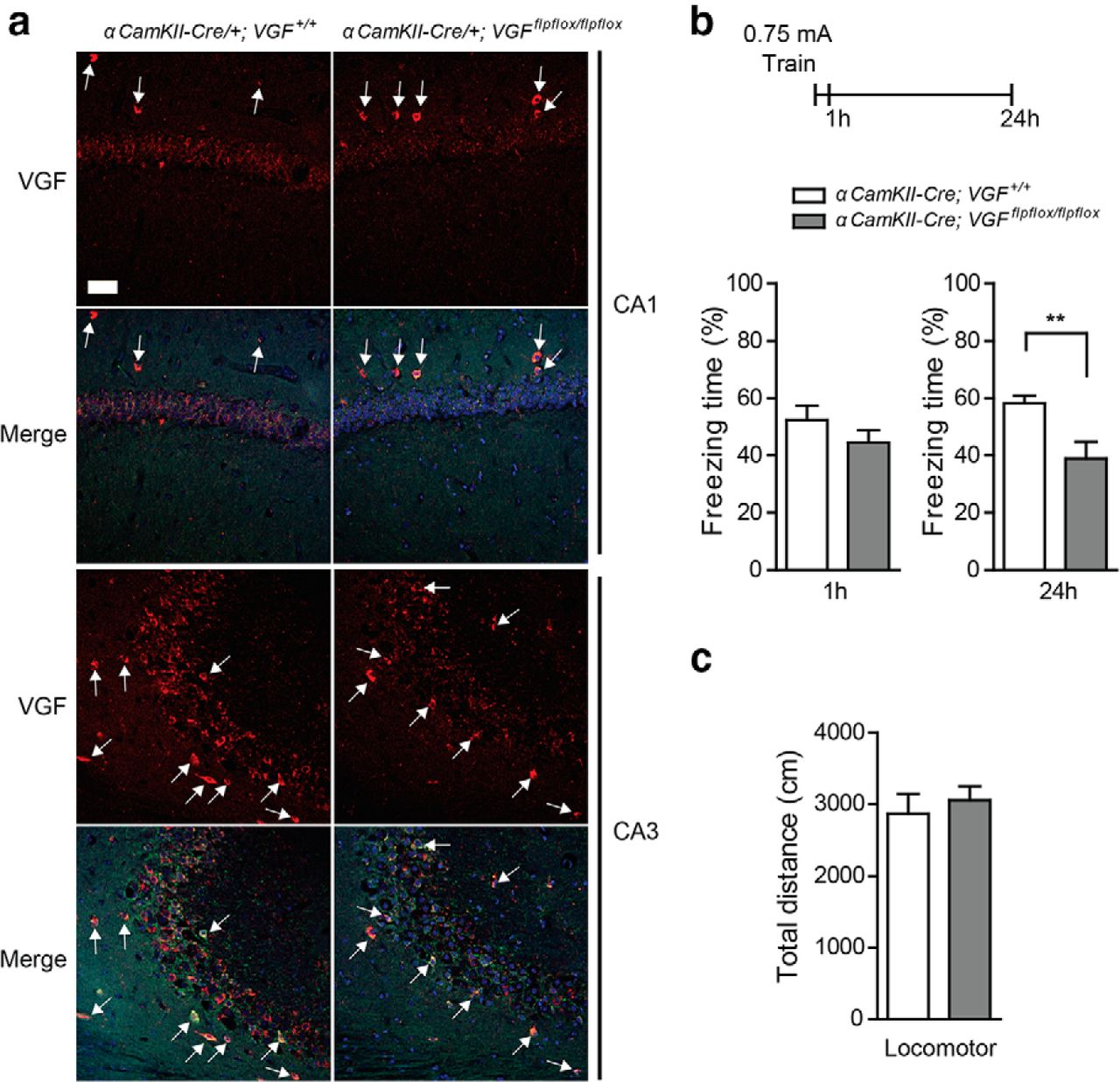
VGF deletion in the forebrain excitatory neurons impairs fear memory formation. a, Brain sections were prepared from αCaMKII-Cre/+;Vgf+/+ and αCaMKII-Cre/+;Vgfflpflox/flpflox mice (5∼7 months old) and stained with rabbit anti-VGF (C-terminal) polyclonal antibody. Decreased VGF staining was observed in the CA1 and CA3 pyramidal neurons of αCaMKII-Cre/+;Vgfflpflox/flpflox mice (red, anti-VGF Ab; green, anti-GAD-65 Ab; blue, DAPI). White arrow indicates VGF/GAD-65-positive cells. Scale bar, 50 μm. b, Long-term (24 h) but not short-term (1 h) fear memory after CFC training was impaired in αCaMKII-Cre/+;Vgfflpflox/flpflox conditional knock-out mice (n = 7∼16 per group). Freezing was observed and measured 1 or 24 h after training. Bar represents average freezing (%) ± SE. Data were analyzed by Student's t test. **p < 0.01. c, Forebrain VGF deletion does not affect locomotor activity (n = 5 per group). Bar represents total distance traveled (in centimeters) ± SE.
Secreted TLQP-62 peptide in adult hippocampus is required for long-term memory formation
Several peptides from the VGF precursor have been identified as regulators of energy homeostasis and neuronal activity in the brain (Bartolomucci et al., 2011; Ferri et al., 2011). Among these peptides, the C-terminal peptide TLQP-62 (VGF aa 556–617) has been shown to potentiate field EPSPs in hippocampal slices and to enhance synaptic activity in cultured hippocampal neurons (Alder et al., 2003; Bartolomucci et al., 2006; Bozdagi et al., 2008). To investigate whether secreted TLQP-62 is required for memory formation, we used an antibody that specifically recognizes this peptide (Chakraborty et al., 2006) to sequester it in vivo. Wild-type mice injected with anti-VGF565–615 into dorsal hippocampus immediately after CFC training showed impaired long-term memory, whereas those injected with control IgG did not (freezing index, IgG: 51.0 ± 5.3%, anti-VGF565–615: 19.0 ± 3.6%, p < 0.001, Student's t test; Fig. 5a). We also tested another memory paradigm, IA, in rats. A similar effect was also observed in rats injected with anti-VGF565–615 antibody immediately after IA training, in which the memory was then tested 2 and 7 d after training and anti-VGF565–615-injected rats showed a severe memory deficit (latency to enter dark chamber, day 2, IgG: 675.1 ± 114.4 s, anti-VGF565–615: 187.8 ± 70.8 s, p < 0.05; day 7, IgG: 698.9 ± 201.1 s, anti-VGF565–615: 149.6 ± 54.7 s, p < 0.05; Fig. 5b). These results indicate that secreted TLQP-62 peptide in adult hippocampus plays a critical role in aversive memory formation, in both mouse and rat models.

Anti-VGF sequestration of secreted VGF C-terminal peptide TLQP-62, but not its shorter derivative TLQP-21, in hippocampus disrupts fear memory retention a, Impaired long-term memory was observed in mice that received anti-VGF C-terminal antibody (anti-VGF565–615 IgG, 0.2 μg/side) injection immediately after CFC training (n = 7∼11 per group). Bar represents average freezing (%) ± SE. Data were analyzed by Student's t test. ***p < 0.001. b, Posttraining injection of anti-VGF565–615 IgG impaired aversive memory in rat (IA). Secreted VGF C-terminal peptides were sequestered by anti-VGF565–615 IgG injection immediately after training (0.2 μg/side, n = 3∼4 per group). Bar represents average latency (sec, for IA) ±SE. Data were analyzed by two-way ANOVA, main effect of treatment, F(1,15) = 13.61, p = 0.0022; main effect of training, F(2,15) = 8.30, p = 0.0037; Bonferroni post hoc test. *p < 0.05. c, Fear memory was selectively altered by anti-AQEE-30 IgG, but not anti-TLQP-21, IgG or control IgG injection (0.5 μg/side, n = 4∼7 per group). Data were analyzed by one-way ANOVA followed by Tukey post hoc test. *p < 0.05. Bar represents average freezing (%) ± SE. d, Specific recognition of cognate peptide AQEE-30 and its parent C-terminal peptide TLQP-62 by anti-AQEE-30 IgG, as revealed by dot blot analysis. Anti-TLQP-21 IgG recognized the cognate TLQP-21 peptide, but not its parent TLQP-62 peptide. e, Limited diffusion of injected IgG in mouse dorsal hippocampus. Brains were stereotactically injected with Alexa Fluor 488-conjugated goat IgG (0.5 μg in 1 μl volume) over 5 min and, after an additional 5 min, mice were anesthetized, perfused, and brains were removed and processed for immunohistochemical staining as described in Materials and Methods. Green, Alexa Fluor 488-conjugated goat IgG; blue, DAPI. Scale bar, 500 μm.
TLQP-62 can be further processed into two shorter peptides, TLQP-21 and AQEE-30, both identified in brain extracts, which could potentially contribute to memory formation (Bartolomucci et al., 2006; Mishiro-Sato et al., 2010). Using specific antibodies raised against TLQP-21 (VGF aa 556–576) or AQEE-30 (VGF aa 588–617), we further examined the requirement for these two shorter processed peptides in contextual fear memory performance. We found that long-term memory formation was blocked in mice injected with anti-AQEE-30 antibody (one-way ANOVA, F(2,12) = 7.06, p = 0.009; Fig. 5c), but not those injected with anti-TLQP-21, which blocks TLQP-21-evoked thermal hyperalgesia but does not recognize native TLQP-62 (Fig. 5d; Fairbanks et al., 2014), nor those injected with control IgG. Given that anti-AQEE-30 antibody also recognizes TLQP-62 (Fig. 5d), these findings suggest that secreted VGF-C-terminal peptides, including TLQP-62 and AQEE-30 but not TLQP-21, are required for the early consolidation phase of memory formation. Antibody delivery was localized to dorsal hippocampus with no detectable spread into adjacent brain regions, as revealed by immunohistochemical staining (Fig. 5e), nor did antibody infusion result in hippocampal damage, which could have impaired memory formation, because mice learned normally when CFC training was received 11 d after anti-AQEE30 antibody infusion (data not shown).
Impaired fear memory formation by acute sequestration of secreted VGF C-terminal peptide in adult hippocampus is rescued by local infusion of BDNF or systemic administration of TrkB agonist 7,8-DHF
We have shown previously that TLQP-62 treatment of hippocampal slices potentiated CA1 field EPSPs, which was further blocked by pretreatment with the Trk tyrosine kinase inhibitor K252a or BDNF scavenger TrkB-Fc, suggesting that BDNF-TrkB signaling is required for TLQP-62 actions (Bozdagi et al., 2008). If activation of BDNF-TrkB signaling pathway is downstream of TLQP-62-mediated actions in memory consolidation, we wondered whether the memory impairment caused by acute sequestration of secreted VGF-C-terminal peptide in adult dorsal hippocampus could be rescued by direct activation of the TrkB receptor. To test this hypothesis, we coinjected BDNF together with anti-VGF polyclonal antibody (anti-AQEE-30) into dorsal hippocampus of adult mice immediately after CFC training. Indeed, we found that impaired long-term memory caused by anti-AQEE-30 antibody was restored to the level similar to control IgG group after BDNF administration (freezing index at 24 h, IgG+PBS: 64.5 ± 4.4%; anti-AQEE-30+PBS: 42.3 ± 5.6%; anti-AQEE-30+BDNF: 66.6 ± 5.2%, one-way ANOVA, F(2,21) = 6.872, p = 0.0051; Fig. 6a). Additional evidence came from the systemic administration of the TrkB receptor agonist 7,8-DHF, which has been shown previously to cross the blood–brain barrier within 2 h after intraperitoneal injection and induce TrkB phosphorylation and downstream signaling in mouse brain regions including cortex and amygdala (Jang et al., 2010; Andero et al., 2011; Choi et al., 2012). We found that 7,8-DHF, injected immediately after CFC training, also rescued memory impairment after intrahippocampal injection of anti-VGF polyclonal antibody (freezing index at 24 h, IgG+DMSO: 54.1 ± 4.7%; anti-VGF565–615+DMSO: 31.4 ± 5.0%; anti-VGF565–615+7,8-DHF: 54.8 ± 6.1%, one-way ANOVA, F(2,26) = 6.128, p = 0.006; Fig. 6b). Consistent with TrkB agonist rescue of memory performance in anti-VGF antibody injected adult mice, 7,8-DHF also increased long-term memory performance of germline VGF heterozygous knock-out mice to the level of wild-type mice, whereas this same dosage of 7,8-DHF did not enhance memory performance in the wild-type mice (freezing index at 24 h, WT+DMSO: 59.8 ± 4.6%, WT+7,8-DHF: 57.4 ± 3.8%, Vgf+/−+DMSO: 19.4 ± 3.8%, Vgf+/−+7,8-DHF: 50.0 ± 4.5%, one-way ANOVA, F(3,31) = 21.87, p < 0.0001; Fig. 6c). Therefore, our results strongly suggest that VGF-mediated function in the dorsal hippocampus during memory consolidation is transduced via TLQP62-mediated activation of BDNF-TrkB signaling pathways.

Acute sequestration of secreted VGF C-terminal peptide in adult hippocampus disrupts fear memory retention, which is rescued by local infusion of BDNF or systemic administration of TrkB agonist 7,8-DHF. a, Impaired contextual fear memory formation by acute dorsal hippocampal injection of anti-VGF C-terminal antibody (anti-AQEE-30, 0.5 μg/side) can be rescued by coinjection with BDNF (0.25 μg/side). Antibodies mixed with PBS or BDNF were coinjected immediately after training. n = 8 per group. b, TrkB agonist 7,8-DHF restored memory deficits caused by anti-VGF C-terminal antibody (anti-VGF565–615, 0.2 μg/side) injection. Immediately after anti-VGF565–615 injection, 7,8-DHF (5 mg/kg) or vehicle was administrated by intraperitoneal injection. n = 8–12 per group. c, Injection of 7,8-DHF immediately after training also improved memory performance in VGF germline heterozygous knock-out mice to the level of wild-type mice, but did not enhance memory when administered to wild-type mice. n = 7∼10 per group. Data were analyzed by one-way ANOVA followed by Tukey post hoc test. *p < 0.05; **p < 0.01; ***p < 0.001. Bar represents average freezing (%) ± SE.
Acute TLQP-62 administration induces phosphorylation of TrkB and CREB and enhances memory formation
Because BDNF/TrkB signaling and the downstream activation of CREB in the hippocampus are required for memory formation (Tyler et al., 2002), we investigated whether TLQP-62 administration also activates these pathways. We found that phosphorylation of CREB (pCREB, Ser-133) was robustly increased in the dorsal hippocampus 30 min after TLQP-62 injection in vivo, similar in magnitude and kinetics to pCREB levels measured after recombinant BDNF administration (Fig. 7a). An acute but transient TrkB receptor activation and the subsequent phosphorylation of CREB were also observed in hippocampal brain slices incubated with TLQP-62, but not its scrambled control peptide SC-62 (pTrkB level at 10 min: TLQP-62: 475.7 ± 98.2%; SC-62: 100.0 ± 5.8%, p < 0.01; pCREB level at 30 min: TLQP-62: 195.9 ± 29.8%; SC-62: 100.0 ± 9.3%, p < 0.05, Student's t test; Fig. 7b), indicating that TrkB receptor activation is downstream of TLQP-62 peptide treatment. Given that enhanced CREB-dependent transcription has been shown previously to enhance long-term memory (Suzuki et al., 2011), we therefore tested the hypothesis that increasing TLQP-62 levels in dorsal hippocampus would enhance memory. Indeed, we found that TLQP-62 administration enhanced long-term memory formation after weak CFC training compared with the control group, which received scrambled peptide SC-62 (freezing index expressed as a percentage of SC-62 control, at 24 h, TLQP-62: 159.1 ± 11.7%; SC-62: 100.0 ± 19.8%, Student's t test, p = 0.02; Fig. 7c). Given that acute administration of anti-AQEE-30, but not anti-TLQP-21, antibody also impaired long-term memory, we further tested whether administration of either AQEE-30 or TLQP-21 into dorsal hippocampus facilitated long-term memory formation. Under weak CFC training, administration of AQEE-30 had no detectable effect, whereas mice that received bilateral injection of TLQP-21 immediately after training had reduced long-term memory, indicating opposing effects of TLQP-21 and its parent peptide TLQP-62 in memory formation (freezing index as percentage of PBS control, at 24 h, PBS: 100.0 ± 6.0%; TLQP-21: 62.5 ± 9.9%; AQEE-30: 111.4 ± 12.7%, one-way ANOVA, F(2,15) = 6.60, p = 0.008; Fig. 7d).

TLQP-62 peptide acutely activates TrkB receptor-mediated signaling pathways and enhances fear memory formation. a, b, VGF C-terminal peptide TLQP-62 enhances BDNF signaling. a, TLQP-62 peptide rapidly induced CREB phosphorylation in vivo. Mouse dorsal hippocampus was injected with the indicated peptide (0.3 μg/side) and tissues were collected 30 min after injection. Immunoblot results showed increased CREB phosphorylation at Ser-133 in both TLQP-62- and BDNF-injected brains. b, TLQP-62 peptide induced BDNF signaling in mouse hippocampal brain slices. Slices were treated with indicated peptides (10 μm) for 10 min or 30 min. n = 3∼6 slices per group. Protein level was analyzed by Student's t test. *p < 0.05; **p < 0.01. c, TLQP-62 peptide facilitated aversive memory formation. A weak training protocol (0.3 mA) was delivered to C57BL/6J mice, which received bilateral hippocampal injections of TLQP-62 or its scrambled control peptide SC-62 (0.5 μg/side) immediately after training. Long-term memory was tested 24 h later. The TLQP-62-injected group showed significant memory enhancement compared with the control group (n = 5∼6 per group). Bar represents the percentage of average freezing compared with SC-62 control group (%) ± SE. Data were analyzed by Student's t test. *p < 0.05. d, Using the same weak training protocol, administration of TLQP-21 peptide reduced long-term fear memory, whereas no effect was detected with AQEE-30. Mice received bilateral hippocampal injections of TLQP-21 (0.5 μg/side), AQEE-30 (0.5 μg/side), or PBS immediately after training and long-term memory was tested 24 h later. The TLQP-21-injected group showed reduced fear memory compared with the PBS control and AQEE-30 groups (n = 6 per group). Data were analyzed by one-way ANOVA followed by Tukey post hoc test. *p < 0.05; **p < 0.01. Bar represents the percentage of average freezing compared with PBS control group (%) ± SE.
VGF regulates training-induced molecular changes associated with actin polymerization in spines and presynaptic release via BDNF-TrkB signaling
Previous studies have shown that BDNF activation of the TrkB receptor and its downstream signaling pathways drives changes in hippocampal synaptic plasticity, including phosphorylation of cofilin and synapsin, changes that are known to regulate postsynaptic cytoskeletal dynamics and presynaptic neurotransmitter release (Jovanovic et al., 2000; Blum and Konnerth, 2005; Rex et al., 2007). We found that phosphorylation of both cofilin and synapsin was increased in the dorsal hippocampus 24 h after CFC training in wild-type (24 h p-cofilin level: 181.8 ± 24.4% compared with naive, p < 0.01; 24 h p-synapsin level: 166.3 ± 16.3% compared with naive, p < 0.01; Fig. 8a), but not in germline VGF heterozygous knock-out mice, indicating a requirement for VGF in training-induced synaptic changes. We also examined the small GTPase Rac1, which is known to activate cofilin phosphorylation and downstream signaling cascades, resulting in dynamic changes in actin polymerization and morphological changes in dendritic spines (Yang et al., 1998; Tashiro et al., 2000). We noted a late-onset Rac1 mRNA induction in the dorsal hippocampus 24 h after fear memory training in wild-type mice (p < 0.001; Fig. 8b), which was absent in VGF heterozygous knock-out mice. Our findings, therefore, indicate a possible role for VGF in the induction and persistence of learning-associated changes in synaptic plasticity. We further investigated whether these changes are mediated by BDNF-TrkB signaling. Indeed, 7,8-DHF injections right after training restored Rac1 mRNA induction in VGF heterozygous knock-out mice 24 h after CFC training (one-way ANOVA, F(2,10) = 4.296, p = 0.045; Fig. 8c). In adult hippocampal slices, incubation of TLQP-62 peptide also rapidly promoted cofilin phosphorylation after 30 min of treatment, whereas total cofilin levels remained unchanged. Importantly, this effect was eliminated by pretreatment with the BDNF scavenger TrkB-Fc (p-cofilin level: SC-62: 100.0 ± 13.3%, TLQP-62: 199.4 ± 25.1%, TrkB-Fc+TLQP-62: 80.5 ± 24.5%, one-way ANOVA, p = 0.0081; Fig. 8d). These results suggest that VGF and its C-terminal TLQP-62 peptide are required for triggering acute and long-lasting cytoskeletal modification in the memory-trained hippocampus through BDNF-activated pathways, which are considered to be critical for establishing the synaptic changes required for the persistence of long-term memory (Tyler et al., 2002; Minichiello, 2009; Deinhardt and Chao, 2014; Lynch et al., 2014).

Dysregulated induction of synaptic plasticity markers in the dorsal hippocampus of fear-memory-trained VGF heterozygous knock-out mice is rescued by systemic administration of the TrkB agonist 7,8-DHF. a, CFC training increased both cofilin and synapsin protein phosphorylation in the dorsal hippocampi of wild-type, but not germline heterozygous Vgf+/− knock-out mice. n = 4∼10 per group. Data were analyzed by Student's t test. **p < 0.01. b, Rac1 mRNA expression was increased in dorsal hippocampus of wild-type mice, but not Vgf+/− mice 24 h after training. n = 3∼9 per group. Data were analyzed by two-way ANOVA, followed by Bonferroni post hoc test. *p < 0.05; ***p < 0.001. c, 7,8-DHF injection increased Rac1 mRNA levels in Vgf+/− heterozygous knock-out mouse hippocampus 24 h after training. n = 4–5 per group. Data were analyzed by one-way ANOVA, followed by Tukey post hoc test. *p < 0.05. d, Acute changes in the phosphorylation level of synaptic plasticity marker cofilin, triggered by TLQP-62 treatment in hippocampal slices, are dependent on the BDNF-TrkB signaling. Hippocampal slices were treated with TLQP-62 or control scrambled peptide SC-62 (10 μm) for 30 min. Changes in cofilin phosphorylation in the TLQP-62-treated group were blocked by TrkB-Fc pretreatment (5 μg/ml). n = 4 per group. Data are analyzed by one-way ANOVA and Tukey post hoc test. *p < 0.05; **p < 0.01.
Discussion
BDNF plays a critical role in cognitive function, including in memory formation (Tyler et al., 2002; Andero et al., 2014; Bekinschtein et al., 2014). We therefore explored the downstream targets of BDNF and potential upstream regulatory mechanisms and identified VGF and its secreted C-terminal peptide TLQP-62 as essential modulators of fear memory consolidation in the adult hippocampus. TLQP-62 induced acute and transient TrkB receptor activation, which in turn regulates memory consolidation. Analysis also revealed an absence of molecular changes in the VGF-deficient hippocampus that are associated with synaptic plasticity and are often regulated through BDNF-TrkB-dependent mechanisms (Jovanovic et al., 2000; Blum and Konnerth, 2005; Rex et al., 2007). Our findings suggest that induction of TLQP-62 peptide by learning in the adult hippocampus reinforces rapid BDNF-TrkB signaling, critically contributing to a positive feedback loop that functions in the early consolidation of memory formation (Bambah-Mukku et al., 2014).
Mice with germline VGF deficiency have memory deficits in CFC and the uncued Morris water maze, two hippocampal-dependent memory paradigms (Bozdagi et al., 2008). Because germline VGF ablation could affect the neural circuitry that regulates memory formation developmentally, we used conditional knock-out approaches to show that VGF expression in the adult hippocampus is required for long-term contextual fear memory (consolidation), but not short-term memory (acquisition). VGF is expressed in excitatory and inhibitory neurons in brain and primary hippocampal neurons (van den Pol et al., 1994; Benson and Salton, 1996), consistent with our findings. αCaMKII-Cre-mediated VGF deletion in postnatal excitatory neurons resulted in impaired long-term memory formation, but left short-term memory intact, similar to AAV-Cre-mediated VGF deletion in the adult dorsal hippocampus. These findings suggest that VGF, expressed by excitatory neurons, is involved in memory formation, but do not rule out a role for abundant VGF that is synthesized in hippocampal inhibitory neurons.
VGF is processed into small peptides that are secreted (Bartolomucci et al., 2011; Ferri et al., 2011), including C-terminal peptides TLQP-62 and its two smaller processed peptides, TLQP-21 and AQEE-30, which have all been identified in brain extracts by Western blot analysis and mass spectrometry (Trani et al., 1995; Bartolomucci et al., 2006; Mishiro-Sato et al., 2010). Data presented here demonstrate that TLQP-62 levels are induced by fear memory training in dorsal hippocampus. By injecting specific peptide-scavenging antibodies into dorsal hippocampus, we showed that extracellular TLQP-62, but not TLQP-21, is required for long-term memory formation. Anti-AQEE-30 antisera most strongly recognize the final ∼20 C-terminal amino acids of VGF (Chakraborty et al., 2006), which are shared by AQEE-30 and TLQP-62. Increased quantitative binding of anti-AQEE30 to immobilized TLQP-62 compared with AQEE-30, in dot blot experiments, could be a consequence of increased retention of TLQP-62 on the dot-blot membrane or a higher binding affinity of anti-AQEE-30 for TLQP-62 compared with AQEE-30, perhaps due to differences in peptide size, charge, or conformation when immobilized. Importantly, these experiments demonstrate that anti-TLQP-21 recognizes TLQP-21, but not AQEE-30 nor TLQP-62, and that anti-AQEE-30 recognizes AQEE-30 and TLQP-62, but not TLQP-21.
We showed previously that TLQP-62, but not TLQP-21 nor AQEE-30, enhanced CA1 field EPSPs in hippocampal slices (Bozdagi et al., 2008). Here, we show that TLQP-62, but not AQEE-30 or TLQP-21, facilitates long-term fear memory formation. Unexpectedly, we also found that TLQP-21 administration impaired fear memory under the same weak training protocol. VGF actions with respect to memory formation and consolidation are therefore likely to depend on the relative levels of its differentially processed peptides and the distribution of VGF peptide receptors.
A receptor for the TLQP-62 peptide remains to be identified. It is unlikely that a recently reported receptor for TLQP-21 (Hannedouche et al., 2013), the complement C3a receptor (C3aR1), and its signaling pathways, are also activated by TLQP-62 based on structure/activity studies of the TLQP-21 peptide (Cero et al., 2014). C3aR1 signaling has been suggested to play a role in hippocampal neurogenesis based on studies in knock-out mice (Rahpeymai et al., 2006), so TLQP-21/C3aR1 signaling could contribute to memory and will need to be further tested, although our studies did not identify an essential role for adult hippocampal TLQP-21 in contextual fear memory formation based on stereotactic infusion of anti-TLQP-21. The contribution to memory formation and consolidation of a second recently identified TLQP-21 receptor that functions in neuropathic pain, the globular heads of the C1q receptor (gC1qR; Chen et al., 2013), remains to be investigated.
Neuronal VGF expression is induced by increased neuronal activity in vivo (Snyder et al., 1998b), cellular depolarization in vitro (Salton et al., 1991; Bonni et al., 1999), and the BDNF signaling pathway in vitro and in vivo (Snyder et al., 1998b; Bonni et al., 1999; Eagleson et al., 2001; Alder et al., 2003; Cazzin et al., 2011). Here, we found increased expression of VGF mRNA and protein in dorsal hippocampus during the early phase of memory consolidation. VGF mRNA induction peaked 1 h after training, likely in response to rapid BDNF-TrkB signaling after memory training (Alonso et al., 2002a; Alonso et al., 2002b) that activated CREB and increased transcription of downstream target genes, including VGF (Salton et al., 2000; Alder et al., 2003; Hunsberger et al., 2007) and BDNF itself (Bambah-Mukku et al., 2014). Very rapid, IA learning-induced BDNF expression leads to persistent activation of CREB and C/EBPβ expression, controlling further bdnf exon IV transcription and mediating memory consolidation (Bambah-Mukku et al., 2014). Although previous studies have reported increased TrkB phosphorylation after extended 2- to 4-week-long TLQP-62 treatment in animals (Lin et al., 2014), could VGF, the expression of which is directly induced by BDNF-TrkB signaling, be involved in the rapid autoregulatory BDNF feedback loop? Regulated secretion of TLQP-62 could trigger additional release of proBDNF and BDNF in an autocrine or paracrine manner (Canossa et al., 1997; Blum and Konnerth, 2005), reinforcing the autoregulatory BDNF loop (Cheng et al., 2011; Bambah-Mukku et al., 2014). Indeed, enhanced potentiation of CA1 field EPSPs in TLQP-62-treated brain slices was blocked by pretreatment with the tissue plasminogen activator (tPA) inhibitor tPA STOP or the BDNF scavenger TrkB-Fc, suggesting that TLQP-62 modulates regulated secretion or processing of pro-BDNF (Bozdagi et al., 2008). VGF-antibody-scavenging experiments demonstrated an early requirement for secreted TLQP-62 in CFC and IA memory consolidation, which was rescued by concurrent local BDNF or systematic TrkB agonist administration immediately after training, suggesting that VGF, and in particular TLQP-62, functions in memory formation by modulating BDNF release, processing, or receptor signaling in a critical time window immediately after memory training. Indeed, administration of the TrkB agonist 7,8-DHF 1 h after training failed to rescue the long-term memory deficit observed in VGF heterozygous knock-out mice (data not shown). Because systemic delivery of 7,8-DHF activates TrkB in the brain 2 h after intraperitoneal injection (Jang et al., 2010) and rapid, local release of BDNF after CFC or IA training is thought to play a critical role in memory formation (Andero et al., 2014; Bekinschtein et al., 2014), our data suggest a critical period within 2–3 h after memory training in which the actions of secreted TLQP-62 and BDNF/TrkB activation are required for memory consolidation.
Although systemic TrkB agonist administration rescued deficits in contextual fear memory formation in germline heterozygous VGF knock-out mice, local infusion of BDNF into the dorsal hippocampus did not (data not shown). Germline Vgf ablation could affect the development of hippocampal circuitry that regulates memory consolidation, so these mice may not respond to intrahippocampal BDNF infusion. Alternatively, activation of BDNF-TrkB-dependent mechanisms in regions other than hippocampus may be required to restore memory.
Changes in synaptic plasticity are required for memory formation (Minichiello, 2009; Lynch et al., 2014). Phosphorylation of cofilin, which regulates actin dynamics, is induced in dendrites after theta-burst stimulation via a BDNF-TrkB-dependent mechanism (Lynch et al., 2014). In addition, activation of Rac1, an upstream factor of cofilin-regulated actin polymerization, is also required for hippocampal-dependent long-term memory formation because inactivation of Rac1 by genetic or pharmacological methods also leads to altered synaptic plasticity and memory impairment (Haditsch et al., 2009; Martinez and Tejada-Simon, 2011). We found that Rac1 mRNA levels and cofilin protein phosphorylation were induced 24 h after contextual fear memory training in the dorsal hippocampus of wild-type mice, but not in germline VGF heterozygous knock-out mice. In addition, TLQP-62 triggered acute cofilin phosphorylation in hippocampal slices via BDNF-dependent signaling. Our data, therefore, suggest a critical role of secreted TLQP-62 peptide in modulating synaptic plasticity through BDNF-TrkB-dependent alterations to the actin cytoskeleton, both in the early (30 min) and late (24 h) phases of contextual fear memory formation because recent studies have demonstrated rapid (30 min) and delayed (12 h) BDNF-dependent phosphorylation of CREB, cofilin, and αCaMKII in response to inhibitory avoidance training (Chen et al., 2012; Bambah-Mukku et al., 2014). Our studies therefore support the hypothesis that a positive feedback loop mediates early consolidation of memory formation: BDNF-TrkB signaling induces expression of VGF and its C-terminal peptide TLQP-62 in the hippocampus after memory training, which acts to reinforce rapid BDNF secretion and/or TrkB signaling in the hippocampus.
Footnotes
This work was supported by the National Institutes of Health (Grants MH086499 and MH083496 to S.R.S., Grant DE021996 to L.V. and S.R.S., Grant MH103455 to O.B., and Grant MH065635 to C.M.A.), the Hope for Depression Research Foundation (S.R.S.), and the Brain and Behavior Research Foundation (S.R.S.). We thank Eric Nestler for providing AAV vectors; Giulio Pasinetti for providing research instruments; Jelle Welagen for discussion; Xiaojing Ye for assistance with rat experiments; and Xiaojing Ye and Gary Philips for insightful comments on the manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Stephen R. Salton, Department of Neuroscience, Box 1639, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, New York, NY 10029. stephen.salton{at}mssm.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}